pia
1.5.5
PIA是用於基於MS的蛋白質推理和鑑定分析的工具箱。
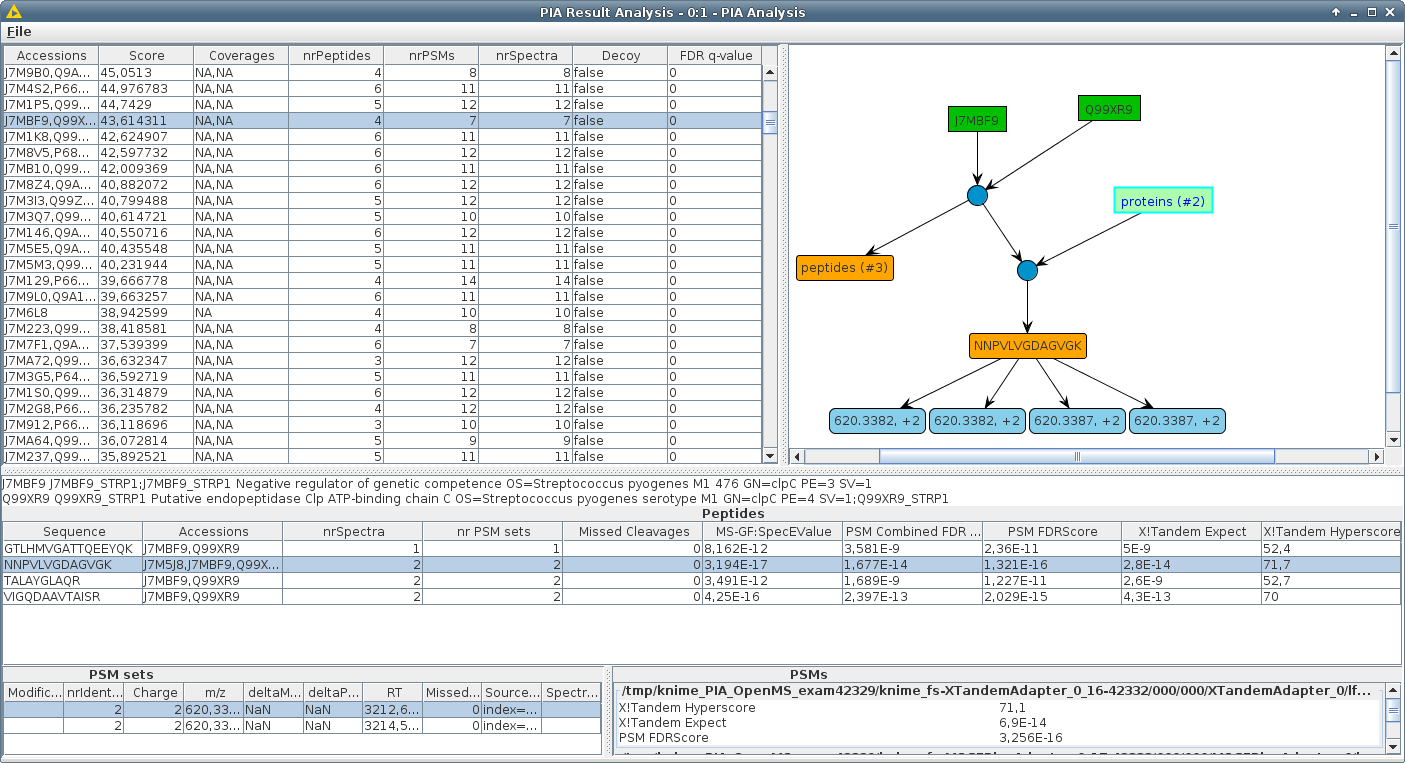
PIA允許您檢查常見的蛋白質組學識別搜索引擎的結果,無縫地組合併進行統計分析。 PIA的主要重點放在綜合的推理算法上,即從一組確定的光譜中得出結論蛋白質。但這也使您可以檢查肽光譜匹配,計算不同搜索引擎結果的FDR值,並可視化PSM,肽和蛋白質之間的對應關係。

儘管實際搜索產生了一組肽光譜匹配(PSMS),但大多數搜索MS/MS實驗中蛋白質識別的引擎都會返回蛋白質列表。從PSM到蛋白質的步驟稱為“蛋白質推理”。如果一組確定的PSM支持在搜索數據庫中檢測多個蛋白質(“蛋白質歧義”),則通常只報告一個代表性登錄。這些代表可能會根據使用的搜索引擎和設置而有所不同。因此,通常無法將不同搜索引擎的蛋白質列表相互比較。互補搜索引擎的PSM通常被合併以增強報告的蛋白質數量或驗證肽的證據,該證據通過使用不同算法的檢測來改善。
我們開發了一個用Java編寫的算法套件,包括完全可參數的knime節點,該節點結合了來自不同實驗和/或搜索引擎的PSM,並報告一致,因此結果一致。沒有像先前的方法那樣固定的參數,例如過濾或評分,但要保持盡可能靈活,以允許用戶需要進行任何調整。
PIA可以通過命令行(也在Docker容器中)或工作流環境knime中調用,這允許無縫集成到OpenMS工作流程中。
對於命令行,您可以使用conda(分別bioconda)下載最新發布的版本,也可以在此處下載構建版本。
PIA也被整合到刀片中。您可以輕鬆地從可信賴的社區貢獻存儲庫中安裝它,該存儲庫都有所有較新的刀具版本。在提交任何錯誤或問題之前,請始終使用最新版本的knime。在Wiki中可以找到有關如何在Knime中找到有關PIA內部PIA的更多信息。
可以在此處下載的教程AS PDF,可在此處找到所需的數據以及此處的工作流(所有數據也可以在https://github.com/mpc-bioinformics/pia-tutorial/的教程存儲庫中可用)。
有關更多文檔,請參閱Github上的Wiki(https://github.com/medbioinf/pia/wiki)。
如果您在PIA有任何問題或查找錯誤和其他問題,請使用GitHub的問題跟踪器(https://github.com/medbioinf/pia/issues)。
如果您發現PIA對您的工作有用,請引用以下出版物:
https://www.ncbi.nlm.nih.gov/pubmed/25938255
https://www.ncbi.nlm.nih.gov/pubmed/30474983
PIA的發展由德國生物信息學基礎設施網絡Elixir / De.NBI資助。
PIA的編程工作由Julian Uszkoreit(Ruhr University Bochum,醫學生物信息學)和Yasset Perez-Riverol(歐洲生物信息學研究所(EMBL-EBI),劍橋)進行


