pia
1.5.5
PIAは、MSベースのタンパク質推論と識別分析のためのツールボックスです。
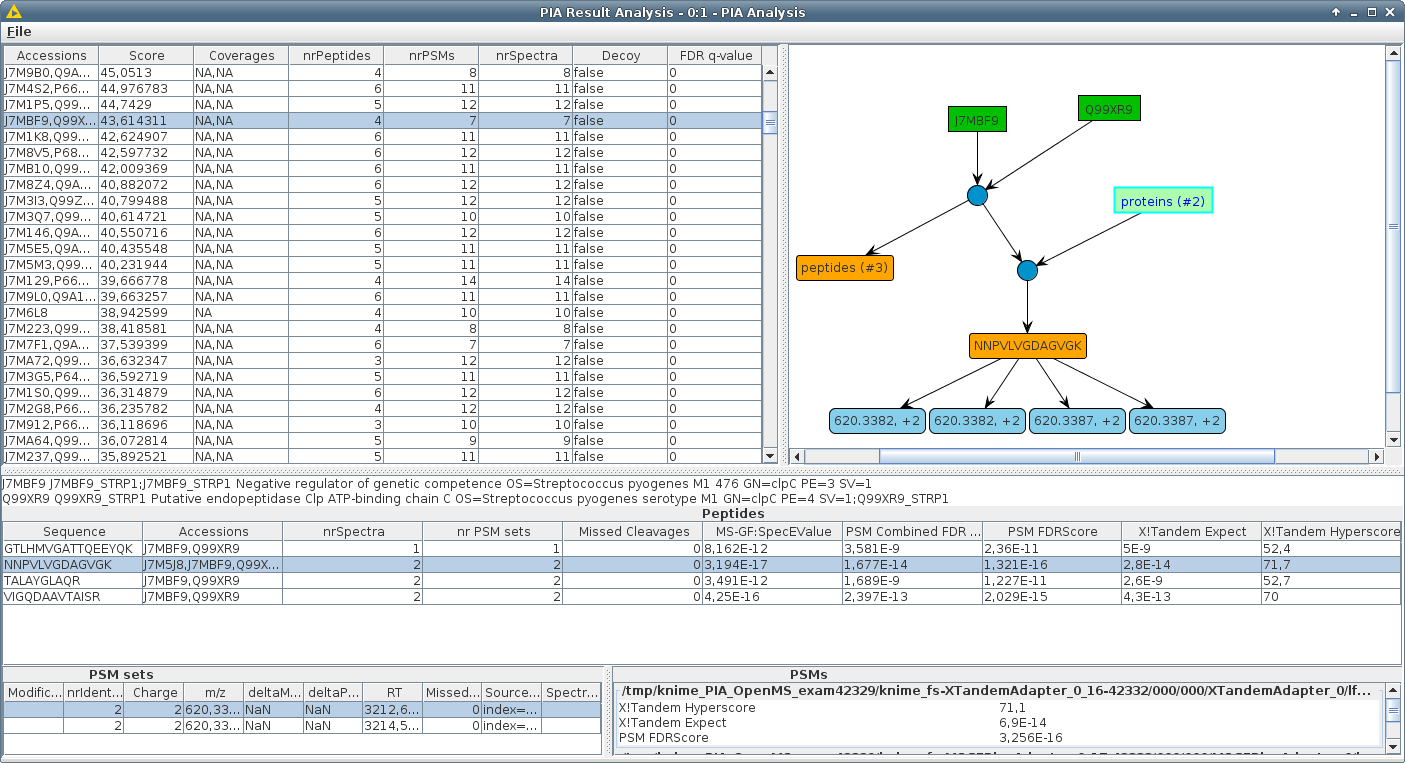
PIAを使用すると、一般的なプロテオミクススペクトル識別検索エンジンの結果を検査し、それらをシームレスに組み合わせて統計分析を実施できます。 PIAの主な焦点は、統合された推論アルゴリズムに基づいています。つまり、特定されたスペクトルのセットからタンパク質を結論付けます。ただし、ペプチドスペクトルの一致を検査し、さまざまな検索エンジンの結果にわたってFDR値を計算し、PSM、ペプチド、タンパク質間の対応を視覚化することもできます。

MS/MS実験でのタンパク質識別のためのほとんどの検索エンジンはタンパク質リストを返しますが、実際の検索ではペプチドスペクトルマッチ(PSM)のセットが得られます。 PSMSからタンパク質へのステップは、「タンパク質推論」と呼ばれます。識別されたPSMのセットが、検索されたデータベース(「タンパク質のあいまいさ」)で複数のタンパク質の検出をサポートする場合、通常1つの代表的なアクセッションのみが報告されます。これらの代表者は、使用済みの検索エンジンと設定によって異なる場合があります。したがって、異なる検索エンジンのタンパク質リストは、一般に互いに比較することはできません。相補的な検索エンジンのPSMは、多くの場合、報告されたタンパク質の数を増やすか、ペプチドの証拠を検証するために組み合わされます。これは、異なるアルゴリズムで検出することで改善されます。
Javaで書かれたアルゴリズムスイートを開発しました。これには、さまざまな実験や検索エンジンのPSMを組み合わせた完全にパラメーション可能なKNIMEノードを含み、一貫した結果と同等の結果を報告しました。フィルタリングやスコアリングなどのパラメーターは、以前のアプローチのように固定されていませんが、ユーザーが必要とする調整を可能にするために、できるだけ柔軟に保持されていません。
PIAは、コマンドライン(Dockerコンテナ)またはワークフロー環境Knimeで呼び出すことができます。これにより、OpenMSワークフローへのシームレスな統合が可能になります。
コマンドラインの場合、Conda(それぞれBioconda)を使用して最新のリリースバージョンをダウンロードするか、ここからビルドをダウンロードできます。
PIAはKnimeにも統合されています。すべての新しいKnimeバージョンで利用できる信頼できるコミュニティの貢献リポジトリから簡単にインストールできます。バグや問題を提出する前に、常に最新バージョンのKnimeを使用してください。 Knime内でPIAをインストールして実行する方法の詳細については、knimeのPIAに関するWikiで見つけることができます。
PDFとしてのチュートリアルはこちらからダウンロードできます。必要なデータはこちらから入手でき、ワークフローはこちらから入手できます(すべてのデータは、https://github.com/mpc-bioinformatics/pia-tutorial/のチュートリアルリポジトリでも入手できます)。
詳細については、githubのwiki(https://github.com/medbioinf/pia/wiki)を参照してください。
PIAに問題がある場合、またはバグやその他の問題を見つけた場合は、GitHub(https://github.com/medbioinf/pia/issues)の問題トラッカーを使用してください。
あなたがあなたの仕事に役立つPIAを見つけた場合、次の出版物を引用してください:
https://www.ncbi.nlm.nih.gov/pubmed/25938255
https://www.ncbi.nlm.nih.gov/pubmed/30474983
PIAの開発は、バイオインフォマティクスインフラストラクチャのためのドイツのネットワークであるElixir / De.nbiによって資金提供されています。
PIAのプログラミング作業は、ジュリアン・ウスコレイト(Ruhr University Bochum、Medical Bioinformatics)、およびYasset Perez-Riverol(European Bioinformatics Institute(EMBL-EBI)、Cambridge)によって行われました。


