pia
1.5.5
PIA ist eine Toolbox für MS -basierte Proteininferenz und Identifikationsanalyse.
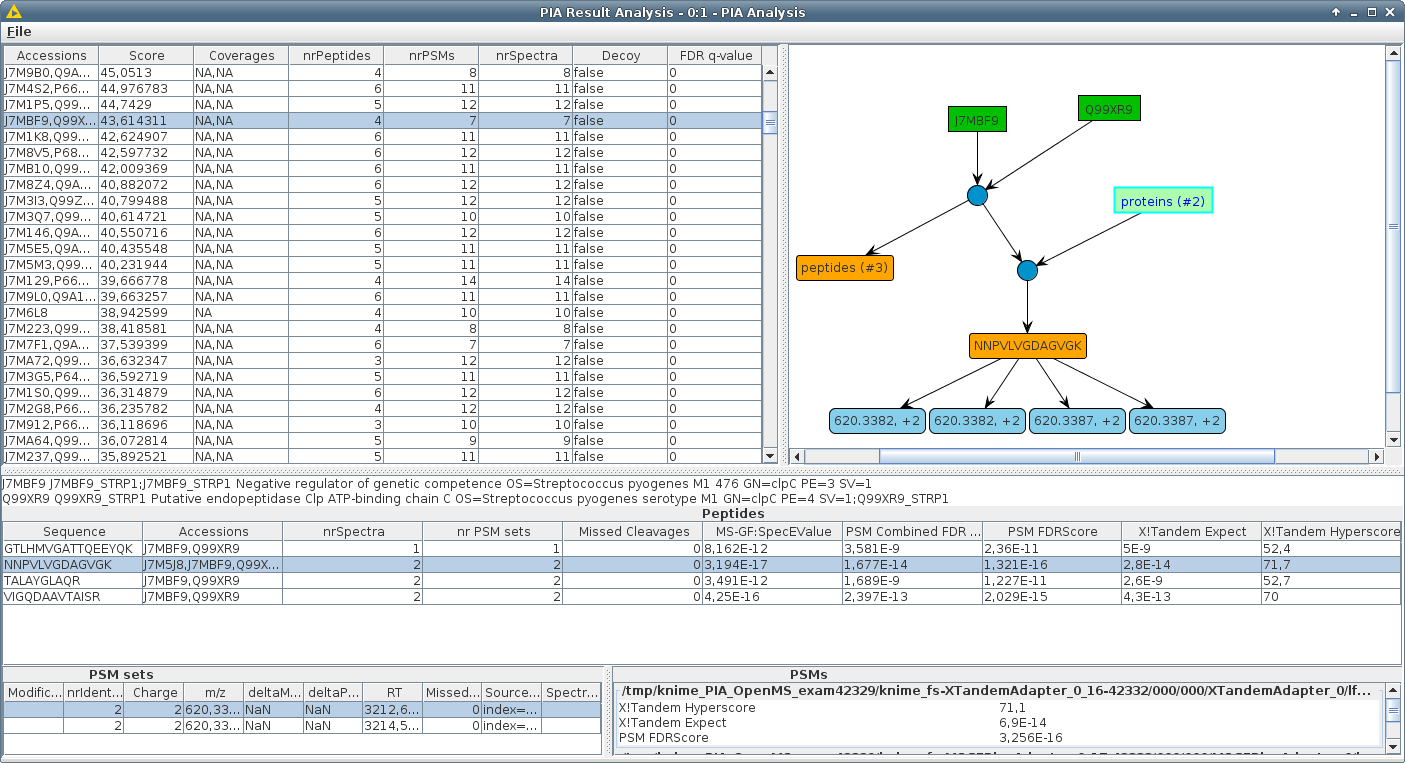
Mit PIA können Sie die Ergebnisse der gängigen Suchmaschinen von Proteomics -Spektrum -Identifikationen überprüfen, sie nahtlos kombinieren und statistische Analysen durchführen. Der Schwerpunkt von PIA liegt auf den integrierten Inferenzalgorithmen, dh den Proteinen aus einer Reihe identifizierter Spektren. Sie können jedoch auch Ihre Peptidspektrum -Übereinstimmungen inspizieren, FDR -Werte über verschiedene Suchmaschinenergebnisse hinweg berechnen und die Korrespondenz zwischen PSMs, Peptiden und Proteinen visualisieren.

Die meisten Suchmaschinen für die Proteinidentifikation in MS/MS -Experimenten geben Proteinlisten zurück, obwohl die tatsächliche Such einen Satz von Peptidspektrum -Übereinstimmungen (PSMs) ergibt. Der Schritt von PSMs zu Proteinen wird als "Proteininferenz" bezeichnet. Wenn ein Satz identifizierter PSMs den Nachweis von mehr als einem Protein in der durchsuchten Datenbank ("Proteinmehrdeutigkeit") unterstützt, wird normalerweise nur ein repräsentativer Zugang gemeldet. Diese Vertreter können sich je nach der verwendeten Suchmaschine und den Einstellungen unterscheiden. Somit können die Proteinlisten verschiedener Suchmaschinen im Allgemeinen nicht miteinander verglichen werden. PSMs komplementärer Suchmaschinen werden häufig kombiniert, um die Anzahl der gemeldeten Proteine zu verbessern oder den Nachweis eines Peptids zu verifizieren, das durch Nachweis mit unterschiedlichen Algorithmen verbessert wird.
Wir haben eine in Java geschriebene Algorithmus -Suite entwickelt, einschließlich vollständig parametrisierbarer Knime -Knoten, die PSMs aus verschiedenen Experimenten und/oder Suchmaschinen kombinieren und konsequent und damit vergleichbare Ergebnisse berichtet. Keiner der Parameter, wie Filterung oder Bewertung, ist wie bei vorherigen Ansätzen festgelegt, aber so flexibel wie möglich, um Anpassungen des Benutzers zu ermöglichen.
PIA kann über die Befehlszeile (auch in Docker -Containern) oder in der Workflow -Umgebung aufgerufen werden, die eine nahtlose Integration in OpenMS -Workflows ermöglicht.
Für die Befehlszeile können Sie die neueste veröffentlichte Version mit Conda (BICONDA) herunterladen oder den Build hier herunterladen.
PIA ist auch in Knime integriert. Sie können es problemlos über das Repository für vertrauenswürdige Community -Beiträge installieren, das in allen neueren Knime -Versionen verfügbar ist. Bitte verwenden Sie immer die neueste Version von Knime, bevor Sie Fehler oder Probleme einreichen. Weitere Informationen zum Installieren und Ausführen von PIA in Knime finden Sie im Wiki über PIA in Knime.
Das Tutorial als PDF kann hier heruntergeladen werden, die erforderlichen Daten sind hier und die Workflows hier verfügbar (alle Daten finden Sie im Tutorial-Repository unter https://github.com/mpc-bioinformatics/pia-tutorial/).
Weitere Unterlagen finden Sie unter GitHub unter dem Wiki (https://github.com/medbioinf/pia/wiki).
Wenn Sie Probleme mit PIA haben oder Fehler und andere Probleme finden, verwenden Sie bitte den Ausgabe -Tracker von GitHub (https://github.com/medbioinf/pia/issues).
Wenn Sie PIA für Ihre Arbeit nützlich fanden, zitieren Sie bitte die folgenden Veröffentlichungen:
https://www.ncbi.nlm.nih.gov/pubmed/25938255
https://www.ncbi.nlm.nih.gov/pubmed/30474983
Die Entwicklung von PIA wird von Elixir / De.NBI, dem deutschen Netzwerk für die Bioinformatik -Infrastruktur, finanziert.
Die Programmierarbeiten über PIA wurden von Julian USzkoreit (Ruhr University Bochum, Medical Bioinformatics) und Yasset Perez-Riverol (Europäisches Bioinformatikinstitut (EMBL-EBI), Cambridge) durchgeführt.


