pia
1.5.5
PIA es una caja de herramientas para la inferencia de proteínas basadas en MS y el análisis de identificación.
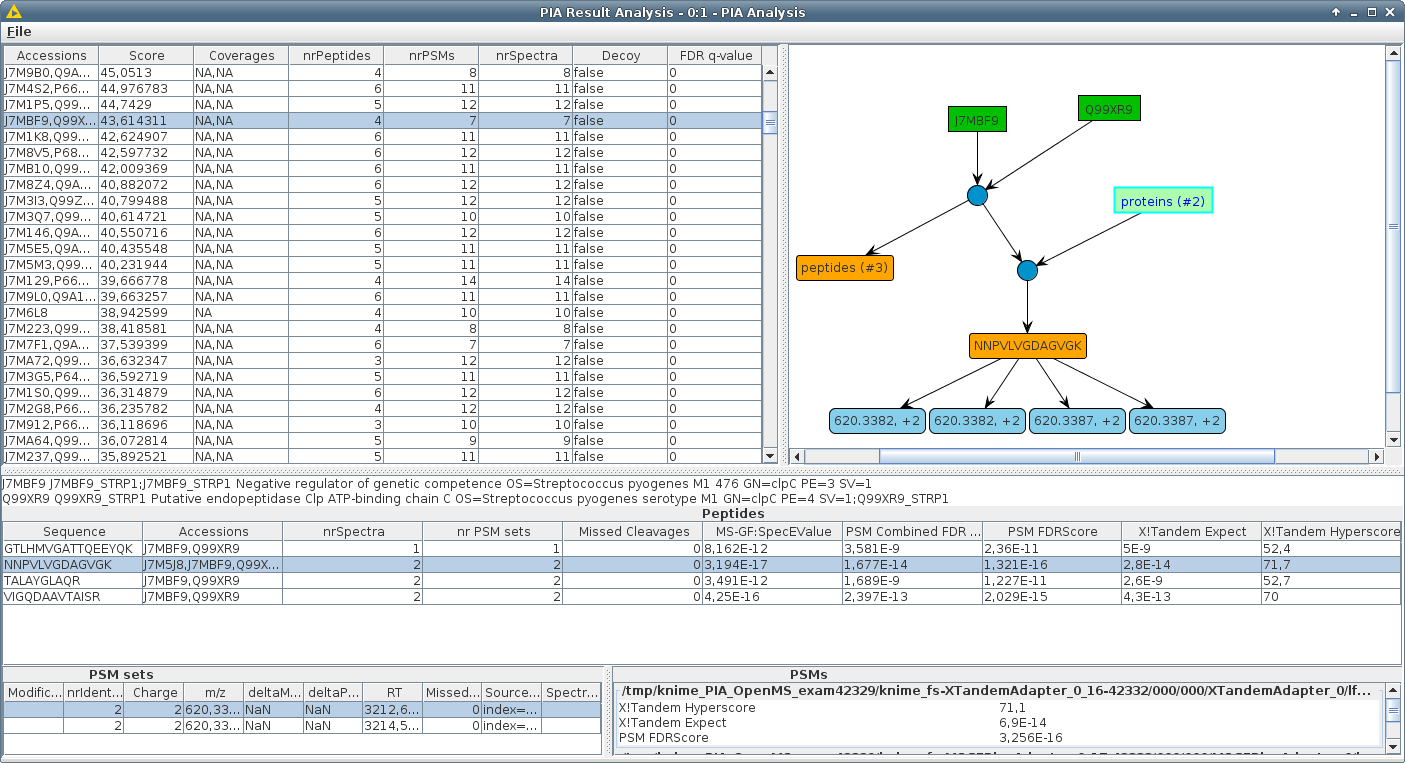
PIA le permite inspeccionar los resultados de los motores de búsqueda de identificación del espectro de proteómica común, combinarlos sin problemas y realizar análisis estadísticos. El enfoque principal de PIA se basa en los algoritmos de inferencia integrados, es decir, concluyendo las proteínas de un conjunto de espectros identificados. Pero también le permite inspeccionar sus coincidencias de espectro de péptidos, calcular los valores de FDR en diferentes resultados de los motores de búsqueda y visualizar la correspondencia entre PSMS, péptidos y proteínas.

La mayoría de los motores de búsqueda para la identificación de proteínas en los experimentos de MS/MS de retorno de listas de proteínas, aunque la búsqueda real produce un conjunto de coincidencias de espectro de péptidos (PSMS). El paso de PSMS a proteínas se llama "inferencia de proteínas". Si un conjunto de PSM identificados admite la detección de más de una proteína en la base de datos buscada ("ambigüedad de proteínas"), generalmente solo se informa una adhesión representativa. Estos representantes pueden diferir de acuerdo con el motor de búsqueda y la configuración usados. Por lo tanto, las listas de proteínas de diferentes motores de búsqueda generalmente no se pueden comparar entre sí. Los PSM de los motores de búsqueda complementarios a menudo se combinan para mejorar el número de proteínas informadas o para verificar la evidencia de un péptido, que se mejora mediante la detección con algoritmos distintos.
Desarrollamos una suite de algoritmo escrita en Java, incluidos nodos de Knime totalmente parametrables, que combinan PSM de diferentes experimentos y/o motores de búsqueda, e informan resultados consistentes y, por lo tanto, resultados comparables. Ninguno de los parámetros, como el filtrado o la puntuación, se soluciona como en enfoques anteriores, pero se mantienen lo más flexibles posible, para permitir cualquier ajuste que el usuario necesita.
Se puede llamar a PIA a través de la línea de comandos (también en contenedores Docker) o en el entorno de flujo de trabajo Knime, lo que permite una integración perfecta en los flujos de trabajo de OpenMS.
Para la línea de comando, puede descargar la última versión lanzada usando Conda (respectivamente BioConda) o descargar la compilación aquí.
PIA también está integrada en Knime. Puede instalarlo fácilmente desde el repositorio de contribuciones de la comunidad de confianza, que está disponible en todas las versiones de Knime más nuevas. Utilice siempre la versión más reciente de Knime antes de enviar cualquier error o problema. Se puede encontrar más información sobre cómo instalar y ejecutar PIA dentro de Knime en el wiki sobre PIA en Knime.
El tutorial como PDF se puede descargar aquí, los datos requeridos están disponibles aquí y los flujos de trabajo aquí (todos los datos también están disponibles en el repositorio del tutorial en https://github.com/mpc-bioinformatics/pia-tutorial/).
Para obtener más documentación, consulte el wiki (https://github.com/medbioinf/pia/wiki) en GitHub.
Si tiene algún problema con PIA o encuentra errores y otros problemas, utilice el rastreador de problemas de GitHub (https://github.com/medbioinf/pia/issues).
Si encontró útil PIA para su trabajo, cite las siguientes publicaciones:
https://www.ncbi.nlm.nih.gov/pubmed/25938255
https://www.ncbi.nlm.nih.gov/pubmed/30474983
El desarrollo de PIA está financiado por Elixir / de.nbi, la red alemana de infraestructura bioinformática.
El trabajo de programación sobre PIA fue realizado por Julian Uszkoreit (Ruhr University Bochum, Medical Bioinformatics) y Yasset Perez-Riverol (Instituto Europeo de Bioinformática (EMBL-EBI), Cambridge)


