pia
1.5.5
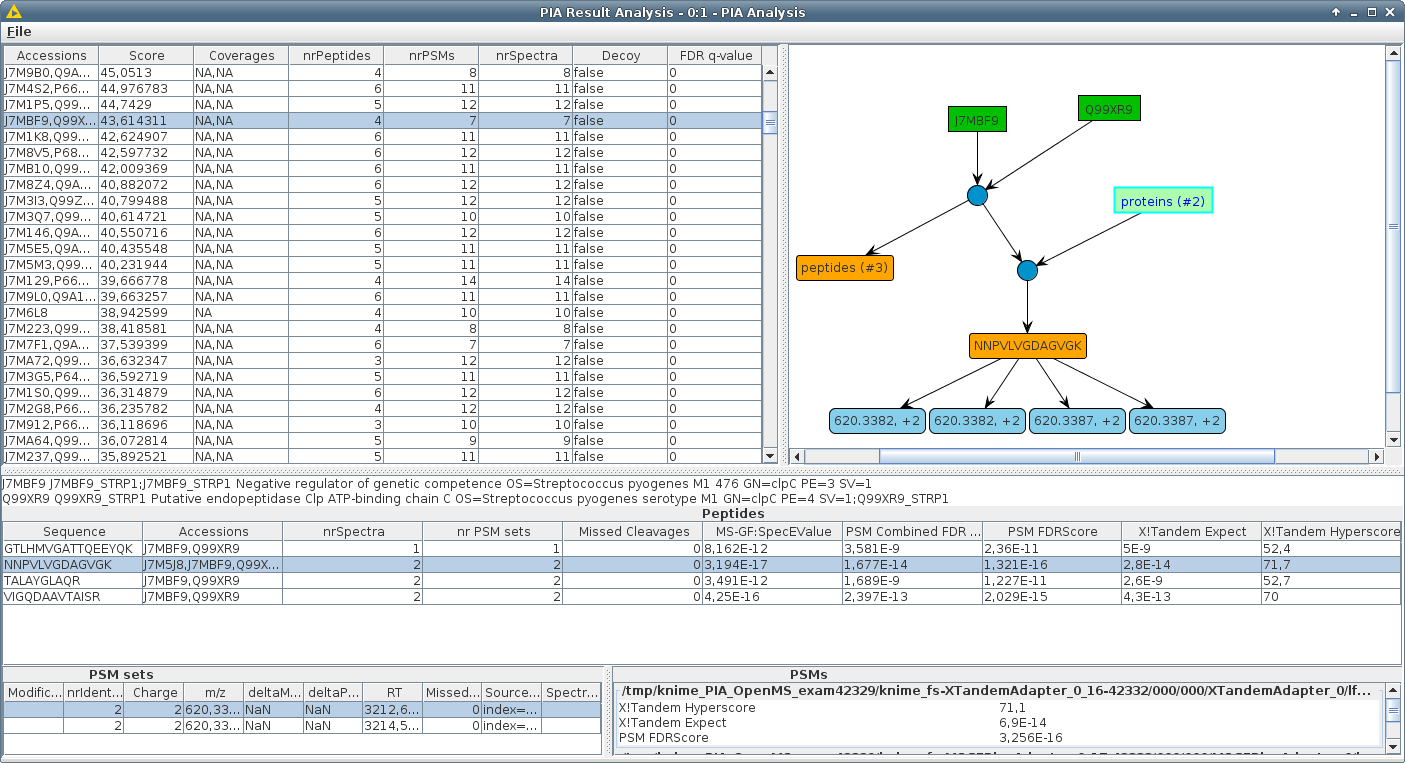
PIA является инструментом для протеинового вывода на основе MS и анализа идентификации.
PIA позволяет вам проверять результаты общих поисковых систем идентификации протеомического спектра, беспрепятственно объединить их и проводить статистический анализ. Основным направлением PIA изложена на интегрированные алгоритмы вывода, т.е. заканчивает белки из набора идентифицированных спектров. Но это также позволяет вам проверять соответствие вашего пептидного спектра, вычислять значения FDR по разным результатам поисковой системы и визуализировать соответствие между PSMS, пептидами и белками.

Большинство поисковых систем для идентификации белка в экспериментах MS/MS возвращают списки белков, хотя фактический поиск дает набор совпадений пептидных спектра (PSMS). Шаг от PSM до белков называется «белковым выводом». Если набор идентифицированных PSM поддерживает обнаружение более чем одного белка в поисковой базе данных («двусмысленность белка»), обычно сообщается только одно репрезентативное вступление. Эти представители могут отличаться в соответствии с использованной поисковой системой и настройками. Таким образом, белковые списки различных поисковых систем обычно не могут сравниваться друг с другом. PSM дополнительных поисковых систем часто объединяются для усиления количества зарегистрированных белков или для проверки доказательств пептида, который улучшается путем обнаружения с помощью отдельных алгоритмов.
Мы разработали набор алгоритмов, написанный на Java, включая полностью параметрируемые узлы ножений, которые объединяют PSM из разных экспериментов и/или поисковых систем, а также согласованные и, следовательно, сопоставимые результаты. Ни один из параметров, таких как фильтрация или оценка, не зафиксирована как в предыдущих подходах, но удерживается как можно более гибкими, чтобы обеспечить любые настройки, необходимые пользователю.
PIA может быть вызвана через командную строку (также в контейнерах Docker) или в ножении среды рабочего процесса, которая обеспечивает бесшовную интеграцию в рабочие процессы OpenMS.
Для командной строки вы можете скачать последнюю выпущенную версию, используя Conda (соответственно BioConda) или загрузить сборку здесь.
PIA также интегрирована в Knime. Вы можете легко установить его из репозитория доверенного сообщества, который доступен во всех более новых ножницах. Пожалуйста, всегда используйте новейшую версию Knime, прежде чем отправлять какие -либо ошибки или проблемы. Более подробную информацию о том, как установить и запустить PIA внутри Knime, можно найти в вики о PIA в Knime.
Учебное пособие как PDF можно загрузить здесь, здесь доступны требуемые данные и рабочие процессы здесь (все данные также доступны в репозитории учебного пособия по адресу https://github.com/mpc-bioinformatics/pia-tutorial/).
Для получения дополнительной документации, пожалуйста, обратитесь к Wiki (https://github.com/medbioinf/pia/wiki) на github.
Если у вас есть какие -либо проблемы с PIA или вы найдете ошибки и другие проблемы, используйте трекер выпуска GitHub (https://github.com/medbioinf/pia/issues).
Если вы нашли PIA полезной для вашей работы, укажите следующие публикации:
https://www.ncbi.nlm.nih.gov/pubmed/25938255
https://www.ncbi.nlm.nih.gov/pubmed/30474983
Разработка PIA финансируется Elixir / DE.NBI, немецкой сети биоинформатики.
Программировая работа по PIA была выполнена Джулианом Ускоретом (Университет Университета Рура, Медицинская биоинформатика) и Яссет Перес-Ривел (Европейский институт биоинформатики (EMBL-EBI), Кембридж)


