pia
1.5.5
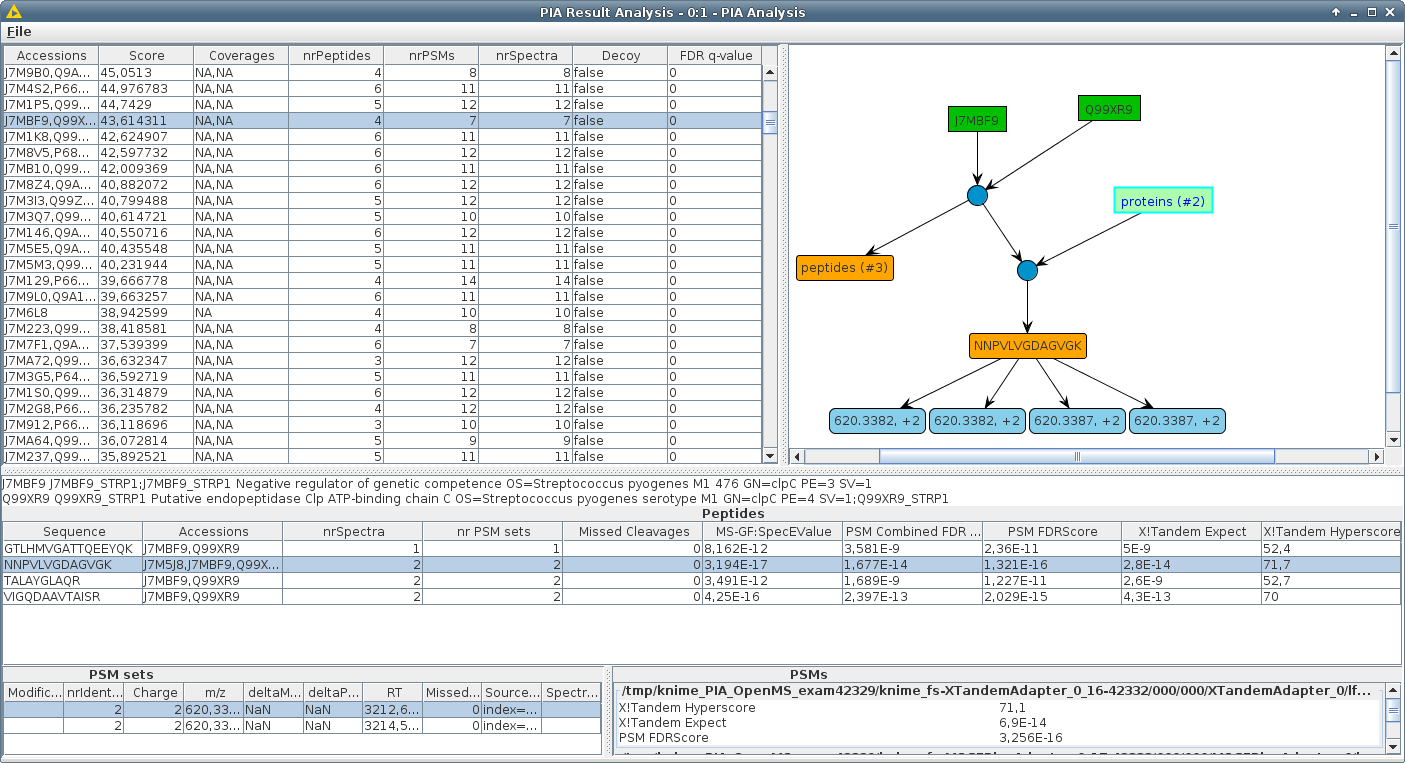
O PIA é uma caixa de ferramentas para análise de inferência e identificação de proteínas baseadas em EM.
A PIA permite inspecionar os resultados dos mecanismos de pesquisa de identificação de espectro proteômico comuns, combiná -los perfeitamente e conduzir análises estatísticas. O foco principal da PIA estabelece os algoritmos de inferência integrada, ou seja, concluindo as proteínas de um conjunto de espectros identificados. Mas também permite que você inspecione as correspondências do espectro de peptídeos, calcule os valores de FDR nos diferentes resultados dos mecanismos de pesquisa e visualize a correspondência entre PSMs, peptídeos e proteínas.

A maioria dos mecanismos de pesquisa para identificação de proteínas em experimentos de MS/MS retornam listas de proteínas, embora a pesquisa real gerem um conjunto de correspondências de espectro peptídico (PSMS). A etapa de PSMs para proteínas é chamada de "inferência de proteína". Se um conjunto de PSMs identificados suportar a detecção de mais de uma proteína no banco de dados pesquisado ("ambiguidade da proteína"), geralmente apenas uma adesão representativa é relatada. Esses representantes podem diferir de acordo com o mecanismo de pesquisa e as configurações usadas. Assim, as listas de proteínas de diferentes mecanismos de pesquisa geralmente não podem ser comparadas entre si. Os PSMs de mecanismos de pesquisa complementares são frequentemente combinados para aprimorar o número de proteínas relatadas ou para verificar a evidência de um peptídeo, que é melhorado pela detecção com algoritmos distintos.
Desenvolvemos um conjunto de algoritmo escrito em Java, incluindo nós de KNIME totalmente parametrisable, que combinam PSMs de diferentes experimentos e/ou mecanismos de pesquisa e relatórios consistentes e, portanto, resultados comparáveis. Nenhum dos parâmetros, como filtragem ou pontuação, é corrigido como em abordagens anteriores, mas mantido o mais flexível possível, para permitir quaisquer ajustes necessários pelo usuário.
O PIA pode ser chamado através da linha de comando (também em contêineres do Docker) ou no ambiente do ambiente de fluxo de trabalho, o que permite uma integração perfeita nos fluxos de trabalho do OpenMS.
Para a linha de comando, você pode baixar a versão mais recente lançada usando o CONDA (respectivamente bioconda) ou baixar a construção aqui.
Pia também é integrado ao KNIME. Você pode instalá -lo facilmente do repositório de contribuições da comunidade confiável, disponível em todas as versões mais recentes do KNIME. Por favor, use sempre a versão mais recente do KNIME antes de enviar bugs ou problemas. Mais informações sobre como instalar e executar PIA dentro do Knime podem ser encontradas no Wiki sobre PIA em Knime.
O tutorial como PDF pode ser baixado aqui, os dados necessários estão disponíveis aqui e os fluxos de trabalho aqui (todos os dados também estão disponíveis no repositório tutorial em https://github.com/mpc-bioinformatics/pia-tutorial/).
Para uma documentação adicional, consulte o Wiki (https://github.com/medbioinf/pia/wiki) no Github.
Se você tiver algum problema com a PIA ou encontrar bugs e outros problemas, use o rastreador de problemas do github (https://github.com/medbioinf/pia/issues).
Se você achou o PIA útil para o seu trabalho, cite as seguintes publicações:
https://www.ncbi.nlm.nih.gov/pubmed/25938255
https://www.ncbi.nlm.nih.gov/pubmed/30474983
O desenvolvimento da PIA é financiado pelo Elixir / de.nbi, a rede alemã para infraestrutura de bioinformática.
O trabalho de programação na PIA foi realizado por Julian Uszkoreit (Universidade Ruhr Bochum, Bioinformática Médica) e Yasset Perez-Riverol (Instituto Europeu de Bioinformática (EMBL-EBI), Cambridge)


