pia
1.5.5
Le PIA est une boîte à outils pour l'inférence et l'analyse d'identification des protéines basées sur la SEP.
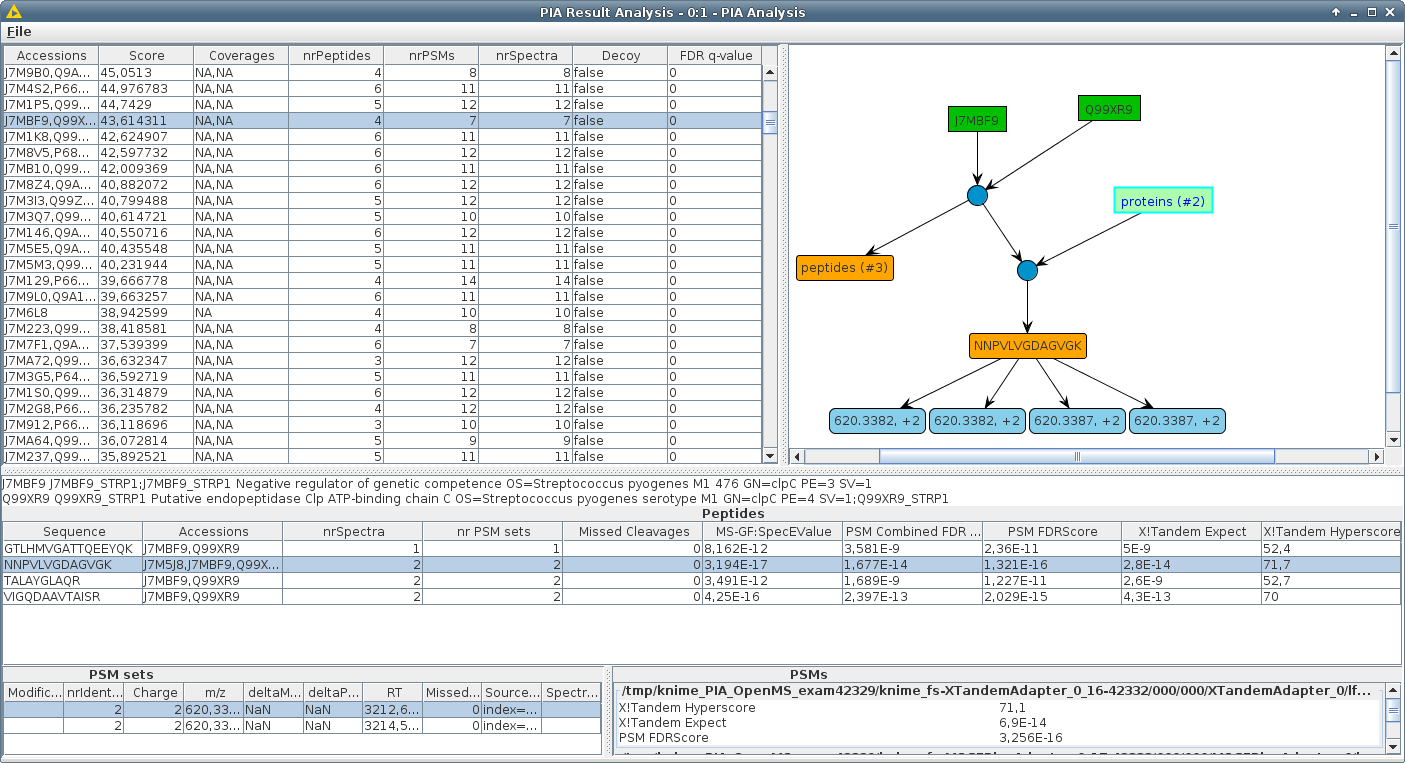
Le PIA vous permet d'inspecter les résultats des moteurs de recherche d'identification du spectre de protéomique commun, de les combiner de manière transparente et d'effectuer des analyses statistiques. L'objectif principal de la PIA est lié aux algorithmes d'inférence intégrés, c'est-à-dire en concluant les protéines d'un ensemble de spectres identifiés. Mais il vous permet également d'inspecter les correspondances du spectre du peptide, de calculer les valeurs FDR à travers différents résultats du moteur de recherche et de visualiser la correspondance entre les PSM, les peptides et les protéines.

La plupart des moteurs de recherche pour l'identification des protéines dans les expériences MS / MS renvoient les listes de protéines, bien que la recherche réelle donne un ensemble de correspondances de spectre peptidique (PSM). L'étape des PSM aux protéines est appelée "inférence des protéines". Si un ensemble de PSM identifiés soutient la détection de plus d'une protéine dans la base de données recherchée ("ambiguïté des protéines"), une seule accession représentative est généralement rapportée. Ces représentants peuvent différer en fonction du moteur de recherche et des paramètres utilisés. Ainsi, les listes de protéines de différents moteurs de recherche ne peuvent généralement pas être comparées les unes aux autres. Les PSM des moteurs de recherche complémentaires sont souvent combinés pour améliorer le nombre de protéines rapportées ou pour vérifier les preuves d'un peptide, qui est amélioré par détection avec des algorithmes distincts.
Nous avons développé une suite d'algorithmes écrite en Java, y compris les nœuds Knime entièrement paramétrisables, qui combinent des PSM à partir de différentes expériences et / ou des moteurs de recherche, et rapporte des résultats cohérents et donc comparables. Aucun des paramètres, comme le filtrage ou la notation, n'est fixé comme dans les approches antérieures, mais maintenus aussi flexibles que possible, pour permettre les ajustements nécessaires à l'utilisateur.
PIA peut être appelé via la ligne de commande (également dans Docker Contaters) ou dans l'environnement de workflow Knime, ce qui permet une intégration transparente dans les flux de travail OpenMS.
Pour la ligne de commande, vous pouvez télécharger la dernière version publiée à l'aide de Conda (respectivement Bioconda) ou télécharger la version ici.
Le PIA est également intégré dans Knime. Vous pouvez facilement l'installer à partir du référentiel de contributions communautaires de confiance, qui est disponible dans toutes les versions KNIME plus récentes. Veuillez utiliser toujours la dernière version de Knime avant de soumettre des bogues ou des problèmes. Plus d'informations sur la façon d'installer et d'exécuter Pia à l'intérieur de Knime peuvent être trouvées dans le wiki sur Pia à Knime.
Le tutoriel en tant que PDF peut être téléchargé ici, les données requises sont disponibles ici et les workflows ici (toutes les données sont également disponibles dans le référentiel de didacticiel à https://github.com/mpc-bioinformatics/pia-tutorial/).
Pour plus de documents, veuillez vous référer au wiki (https://github.com/medbioinf/pia/wiki) sur github.
Si vous avez des problèmes avec PIA ou trouvez des bogues et d'autres problèmes, veuillez utiliser le tracker de problème de GitHub (https://github.com/medbioinf/pia/issues).
Si vous avez trouvé PIA utile pour votre travail, veuillez citer les publications suivantes:
https://www.ncbi.nlm.nih.gov/pubmed/25938255
https://www.ncbi.nlm.nih.gov/pubmed/30474983
Le développement de la PIA est financé par Elixir / de.NBI, le réseau allemand pour l'infrastructure bioinformatique.
Le travail de programmation sur la PIA a été réalisé par Julian Uszkoreit (Ruhr University Bochum, Medical Bioinformatics) et Yasset Perez-Riverol (European Bioinformatics Institute (EMBL-EBI), Cambridge)


