clinker
v0.0.30
現在可以在CageCat Web服務器上安裝CBLASTER和CRENKER。
基因簇比較圖生成器
熟料是一種容易生成出版物質量基因簇比較圖的管道。
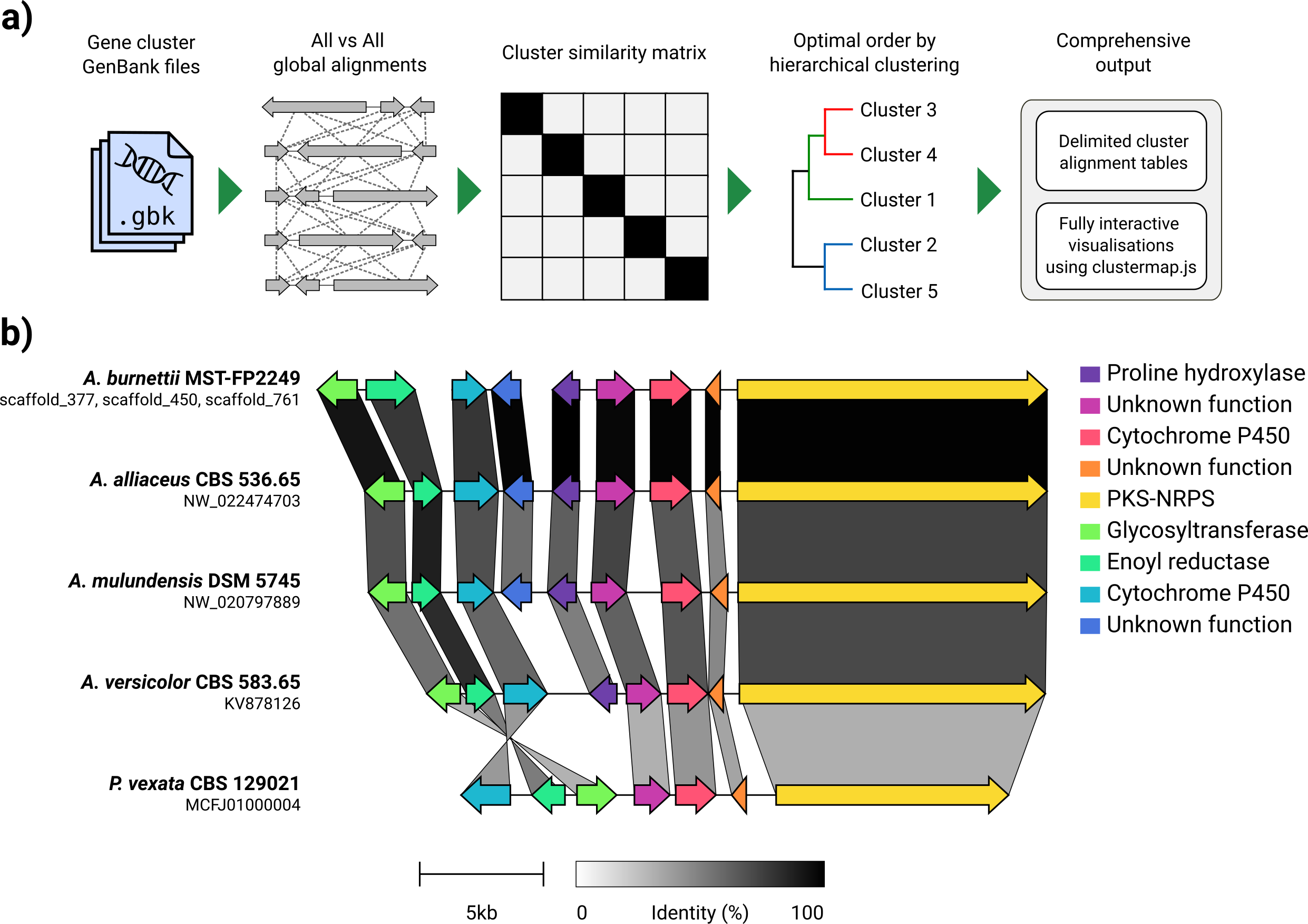
給定一組GenBank文件,熟料將自動提取蛋白質翻譯,在每個群集中的序列之間執行全局對齊,確定基於群集相似性的最佳顯示順序,並生成可以在以前進行廣泛調整的交互式可視化(使用clustermmap.js)被導出為SVG文件。
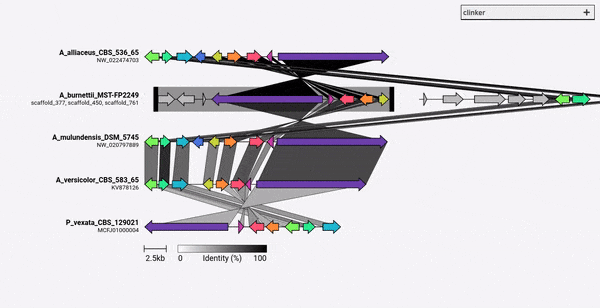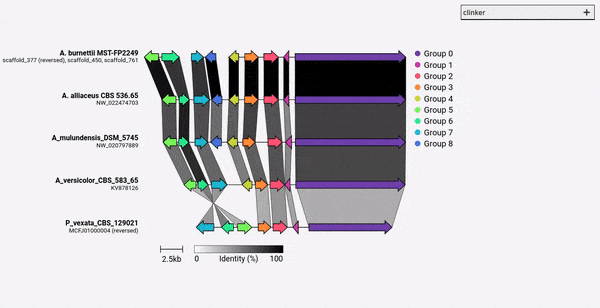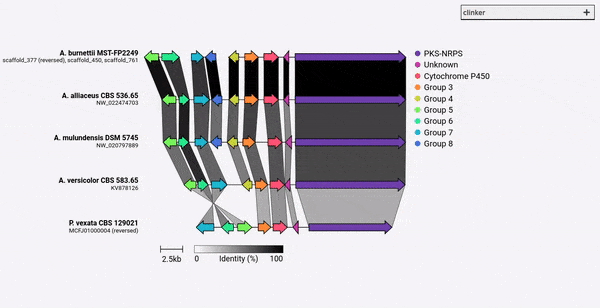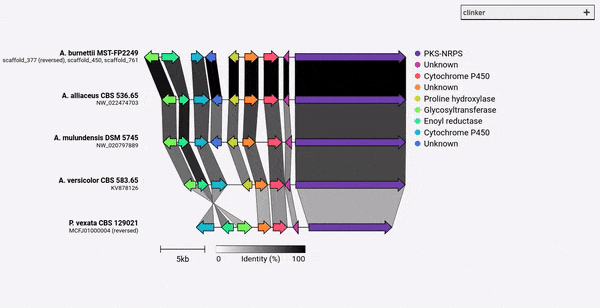
熟料的設計主要是一種可視化同源生物合成基因簇的組的簡單方法,這些基因簇通常是很小的基因組區域,其基因不多(如示例GIF中)。它使用Biopython內置的對齊器在所有輸入文件中執行所有基因的成對對齊,然後在瀏覽器中生成一個交互式SVG文檔。對齊階段的比對階段的縮放範圍很差,可用於多個基因的多個基因組,並且鑑於其將包含多少個SVG元素,所得的可視化也將非常慢。如果您希望將整個基因組保持一致,則使用為此目的構建的工具(例如仙人掌),您可能會更好地為您服務。
熟料可以通過PIP直接安裝:
pip install clinker
通過從github克隆源代碼:
git clone https://github.com/gamcil/clinker.git
cd clinker
pip install .
或者,通過conda:
conda create -n clinker -c conda-forge -c bioconda clinker-py
conda activate clinker
如果您發現熟料有用,請引用:
clinker & clustermap.js: Automatic generation of gene cluster comparison figures.
Gilchrist, C.L.M., Chooi, Y.-H., 2020.
Bioinformatics. doi: https://doi.org/10.1093/bioinformatics/btab007
運行熟料可以很簡單:
clinker clusters/*.gbk
這將在文件夾中的所有GenBank文件中讀取,對齊它們,並將對齊方式打印到終端。要生成可視化,請使用-p/--plot參數:
clinker clusters/*.gbk -p <optional: file name to save static HTML>
熟料還可以解析GFF3文件:
clinker cluster1.gff3 cluster2.gff3 -p
注意:必須在同一目錄中找到相同名稱的相應fasta文件(擴展名為“ .fa”,“ .fsa”,“ .fna”,“ .fasta”或“ cluster1.fa和cluster2.fa 。
請參閱-h/--help有關更多信息:
usage: clinker [-h] [--version] [-r RANGES [RANGES ...]] [-gf GENE_FUNCTIONS] [-na] [-i IDENTITY] [-j JOBS] [-s SESSION] [-ji JSON_INDENT] [-f] [-o OUTPUT] [-p [PLOT]] [-dl DELIMITER] [-dc DECIMALS] [-hl] [-ha] [-mo MATRIX_OUT] [-ufo] [files ...]
clinker: Automatic creation of publication-ready gene cluster comparison figures.
clinker generates gene cluster comparison figures from GenBank files. It performs pairwise local or global alignments between every sequence in every unique pair of clusters and generates interactive, to-scale comparison figures using the clustermap.js library.
optional arguments:
-h, --help show this help message and exit
--version show program's version number and exit
Input options:
files Gene cluster GenBank files
-r RANGES [RANGES ...], --ranges RANGES [RANGES ...]
Scaffold extraction ranges. If a range is specified, only features within the range will be extracted from the scaffold. Ranges should be formatted like: scaffold:start-end (e.g. scaffold_1:15000-40000)
-gf GENE_FUNCTIONS, --gene_functions GENE_FUNCTIONS
2-column CSV file containing gene functions, used to build gene groups from same function instead of sequence similarity (e.g. GENE_001,PKS-NRPS).
Alignment options:
-na, --no_align Do not align clusters
-i IDENTITY, --identity IDENTITY
Minimum alignment sequence identity [default: 0.3]
-j JOBS, --jobs JOBS Number of alignments to run in parallel (0 to use the number of CPUs) [default: 0]
Output options:
-s SESSION, --session SESSION
Path to clinker session
-ji JSON_INDENT, --json_indent JSON_INDENT
Number of spaces to indent JSON [default: none]
-f, --force Overwrite previous output file
-o OUTPUT, --output OUTPUT
Save alignments to file
-p [PLOT], --plot [PLOT]
Plot cluster alignments using clustermap.js. If a path is given, clinker will generate a portable HTML file at that path. Otherwise, the plot will be served dynamically using Python's HTTP server.
-dl DELIMITER, --delimiter DELIMITER
Character to delimit output by [default: human readable]
-dc DECIMALS, --decimals DECIMALS
Number of decimal places in output [default: 2]
-hl, --hide_link_headers
Hide alignment column headers
-ha, --hide_aln_headers
Hide alignment cluster name headers
-mo MATRIX_OUT, --matrix_out MATRIX_OUT
Save cluster similarity matrix to file
Visualisation options:
-ufo, --use_file_order
Display clusters in order of input files
Example usage
-------------
Align clusters, plot results and print scores to screen:
$ clinker files/*.gbk
Only save gene-gene links when identity is over 50%:
$ clinker files/*.gbk -i 0.5
Save an alignment session for later:
$ clinker files/*.gbk -s session.json
Save alignments to file, in comma-delimited format, with 4 decimal places:
$ clinker files/*.gbk -o alignments.csv -dl "," -dc 4
Generate visualisation:
$ clinker files/*.gbk -p
Save visualisation as a static HTML document:
$ clinker files/*.gbk -p plot.html
Cameron Gilchrist, 2020
默認情況下,熟料會自動為每組同源基因分配名稱和顏色。您可以使用-gf/--gene_functions參數預先分配名稱(IE函數),該參數採用2列逗號分隔的文件,例如:
GENE_001,Cytochrome P450
GENE_002,Cytochrome P450
GENE_003,Methyltransferase
GENE_004,Methyltransferase
這將產生兩組,分別是細胞色素P450(Gene_001和002)和甲基轉移酶(Gene_003,Gene_004)。如果發現其他同源基因,它們將自動添加到這些組中。
從熟料v0.0.28開始,您現在可以指定由-gf/--gene_functions參數定義的基因的顏色。為此,請使用-cm/--colour_map參數,該參數還採用一個包含組名稱和十六進制顏色代碼的2個CSV文件,例如:
Cytochrome P450,#FF0000
Methyltransferase,#0000FF


