clinker
v0.0.30
يمكن الآن استخدام كل من Cblaster و Clinker دون تثبيت على خادم الويب Cagecat.
مولد رقم مجموعة مجموعة الجينات
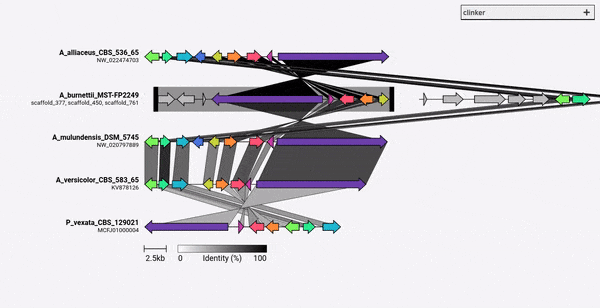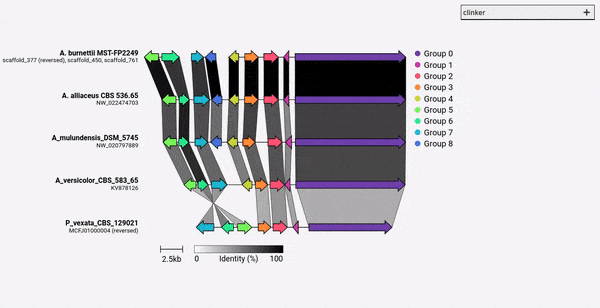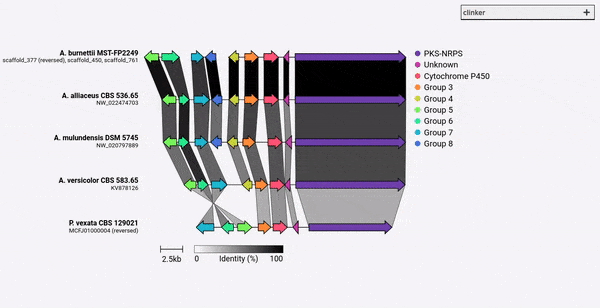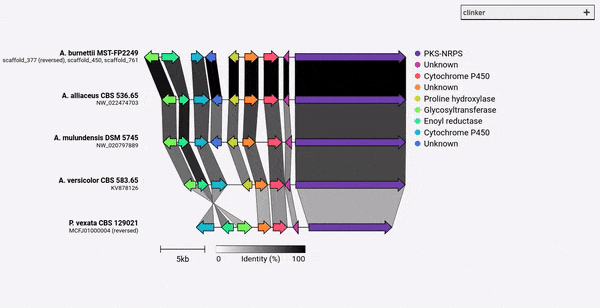
Clinker هو خط أنابيب لتوليد بسهولة أرقام مقارنة مجموعة الجينات جودة الجينات.
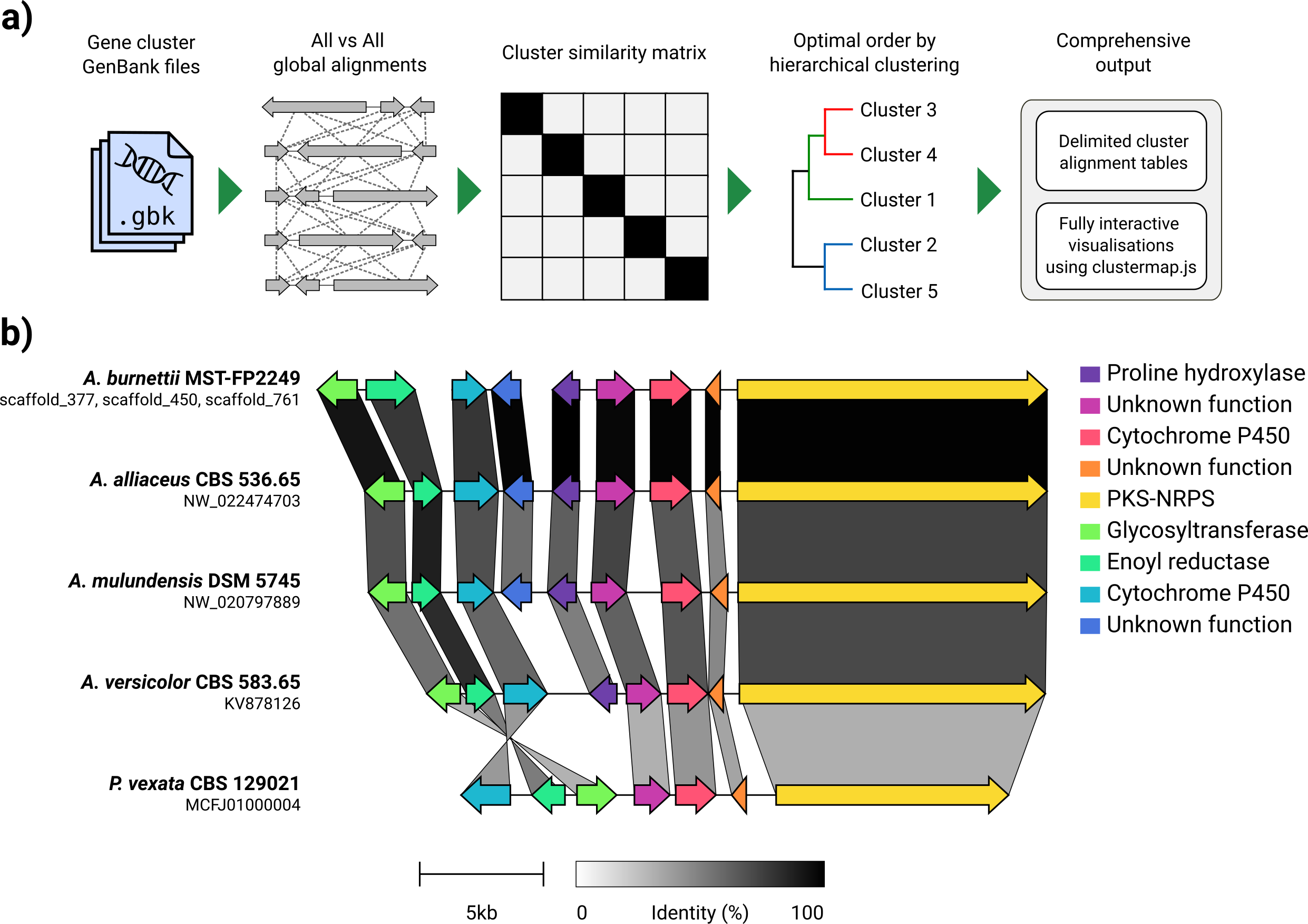
بالنظر إلى مجموعة من ملفات GenBank ، سيقوم Clinker تلقائيًا باستخراج ترجمات البروتين ، وتنفيذ محاذاة عالمية بين التسلسلات في كل مجموعة ، وتحديد ترتيب العرض الأمثل استنادًا إلى تشابه الكتلة ، وإنشاء تصور تفاعلي (باستخدام clustermap.js) يمكن أن يتم تعديله على نطاق واسع من قبل يجري تصديرها كملف SVG.
تم تصميم Clinker في المقام الأول كوسيلة بسيطة لتصور مجموعات من مجموعات الجينات الحيوية المتجانسة ، والتي عادة ما تكون مناطق جينية صغيرة مع عدم وجود العديد من الجينات (كما في مثال GIF). إنه يقوم بمحاذاة زوجية لجميع الجينات في جميع ملفات الإدخال باستخدام جهاز Aligner المدمج في Biopython ، ثم يقوم بإنشاء مستند SVG تفاعلي في المتصفح. ستتوسع مرحلة المحاذاة بشكل سيء للغاية مع جينومات متعددة مع العديد من الجينات ، وسيكون التصور الناتج بطيئًا للغاية نظرًا لعدد عناصر SVG التي سيحتوي عليها. إذا كنت تتطلع إلى محاذاة الجينوم بأكمله ، فمن المحتمل أن يتم تقديمه بشكل أفضل باستخدام الأدوات المصممة لهذا الغرض (مثل الصبار).
يمكن تثبيت Clinker مباشرة من خلال PIP:
pip install clinker
عن طريق استنساخ الكود المصدر من جيثب:
git clone https://github.com/gamcil/clinker.git
cd clinker
pip install .
أو من خلال كوندا:
conda create -n clinker -c conda-forge -c bioconda clinker-py
conda activate clinker
إذا وجدت Clinker مفيدًا ، يرجى الاستشهاد:
clinker & clustermap.js: Automatic generation of gene cluster comparison figures.
Gilchrist, C.L.M., Chooi, Y.-H., 2020.
Bioinformatics. doi: https://doi.org/10.1093/bioinformatics/btab007
يمكن أن يكون تشغيل Clinker بسيطًا مثل:
clinker clusters/*.gbk
سيقرأ هذا في جميع ملفات GenBank داخل المجلد ، ومواءمتها ، وطباعة المحاذاة إلى المحطة. لتوليد التصور ، استخدم وسيطة -p/--plot :
clinker clusters/*.gbk -p <optional: file name to save static HTML>
يمكن لـ Clinker أيضًا تحليل ملفات GFF3:
clinker cluster1.gff3 cluster2.gff3 -p
ملاحظة: يجب العثور على ملف fasta المقابل الذي يحمل نفس الاسم (الامتدادات ".fa" أو cluster1.fa و cluster2.fa .
انظر -h/--help لمزيد من المعلومات:
usage: clinker [-h] [--version] [-r RANGES [RANGES ...]] [-gf GENE_FUNCTIONS] [-na] [-i IDENTITY] [-j JOBS] [-s SESSION] [-ji JSON_INDENT] [-f] [-o OUTPUT] [-p [PLOT]] [-dl DELIMITER] [-dc DECIMALS] [-hl] [-ha] [-mo MATRIX_OUT] [-ufo] [files ...]
clinker: Automatic creation of publication-ready gene cluster comparison figures.
clinker generates gene cluster comparison figures from GenBank files. It performs pairwise local or global alignments between every sequence in every unique pair of clusters and generates interactive, to-scale comparison figures using the clustermap.js library.
optional arguments:
-h, --help show this help message and exit
--version show program's version number and exit
Input options:
files Gene cluster GenBank files
-r RANGES [RANGES ...], --ranges RANGES [RANGES ...]
Scaffold extraction ranges. If a range is specified, only features within the range will be extracted from the scaffold. Ranges should be formatted like: scaffold:start-end (e.g. scaffold_1:15000-40000)
-gf GENE_FUNCTIONS, --gene_functions GENE_FUNCTIONS
2-column CSV file containing gene functions, used to build gene groups from same function instead of sequence similarity (e.g. GENE_001,PKS-NRPS).
Alignment options:
-na, --no_align Do not align clusters
-i IDENTITY, --identity IDENTITY
Minimum alignment sequence identity [default: 0.3]
-j JOBS, --jobs JOBS Number of alignments to run in parallel (0 to use the number of CPUs) [default: 0]
Output options:
-s SESSION, --session SESSION
Path to clinker session
-ji JSON_INDENT, --json_indent JSON_INDENT
Number of spaces to indent JSON [default: none]
-f, --force Overwrite previous output file
-o OUTPUT, --output OUTPUT
Save alignments to file
-p [PLOT], --plot [PLOT]
Plot cluster alignments using clustermap.js. If a path is given, clinker will generate a portable HTML file at that path. Otherwise, the plot will be served dynamically using Python's HTTP server.
-dl DELIMITER, --delimiter DELIMITER
Character to delimit output by [default: human readable]
-dc DECIMALS, --decimals DECIMALS
Number of decimal places in output [default: 2]
-hl, --hide_link_headers
Hide alignment column headers
-ha, --hide_aln_headers
Hide alignment cluster name headers
-mo MATRIX_OUT, --matrix_out MATRIX_OUT
Save cluster similarity matrix to file
Visualisation options:
-ufo, --use_file_order
Display clusters in order of input files
Example usage
-------------
Align clusters, plot results and print scores to screen:
$ clinker files/*.gbk
Only save gene-gene links when identity is over 50%:
$ clinker files/*.gbk -i 0.5
Save an alignment session for later:
$ clinker files/*.gbk -s session.json
Save alignments to file, in comma-delimited format, with 4 decimal places:
$ clinker files/*.gbk -o alignments.csv -dl "," -dc 4
Generate visualisation:
$ clinker files/*.gbk -p
Save visualisation as a static HTML document:
$ clinker files/*.gbk -p plot.html
Cameron Gilchrist, 2020
بشكل افتراضي ، يقوم Clinker تلقائيًا بتعيين اسم ولون لكل مجموعة من الجينات المتماثلة. يمكنك بدلاً من ذلك الحصول على أسماء مسبقة (وظائف IE) باستخدام وسيطة -gf/--gene_functions ، والتي تأخذ ملفًا مفصلاً من عمود الفاصلة مثل:
GENE_001,Cytochrome P450
GENE_002,Cytochrome P450
GENE_003,Methyltransferase
GENE_004,Methyltransferase
سيؤدي ذلك إلى إنشاء مجموعتين ، السيتوكروم P450 (Gene_001 و 002) ، و Methyltransferase (Gene_003 ، Gene_004). إذا تم تحديد أي جينات متماثلة أخرى ، فسيتم إضافتها تلقائيًا إلى هذه المجموعات.
اعتبارًا من Clinker V0.0.28 ، يمكنك الآن تحديد الألوان للجينات المحددة بواسطة وسيطة -gf/--gene_functions . للقيام بذلك ، استخدم وسيطة -cm/--colour_map التي تأخذ أيضًا ملف CSV من عمود يحتوي على اسم المجموعة ورمز اللون السداسي الشريسي مثل:
Cytochrome P450,#FF0000
Methyltransferase,#0000FF


