clinker
v0.0.30
CBLASTER et CLINKER peuvent désormais être utilisés sans installation sur le serveur Web Cagecat.
Génique de comparaison du cluster Générateur
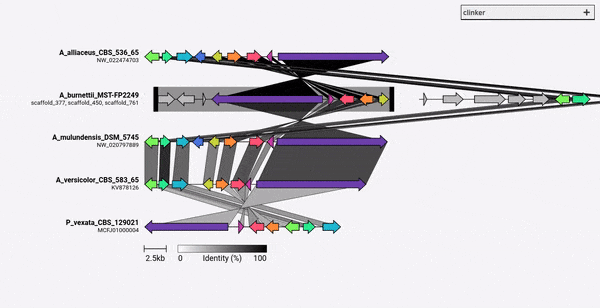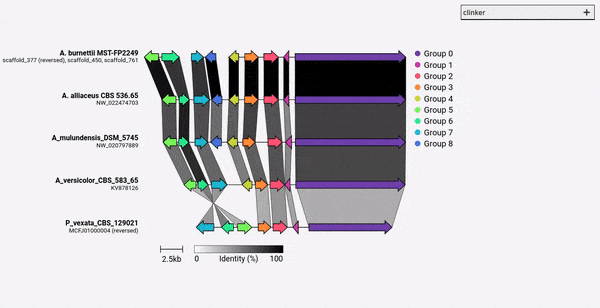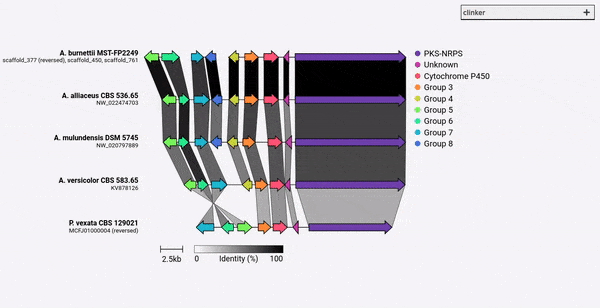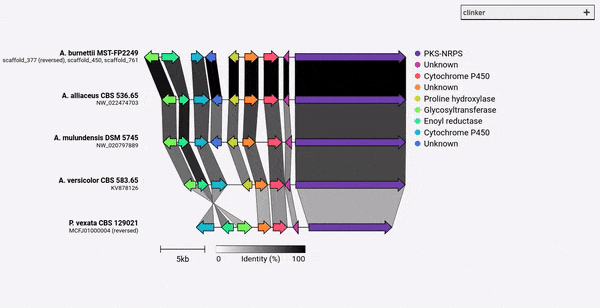
Clinker est un pipeline pour générer facilement des chiffres de comparaison de cluster de gènes de qualité publication.
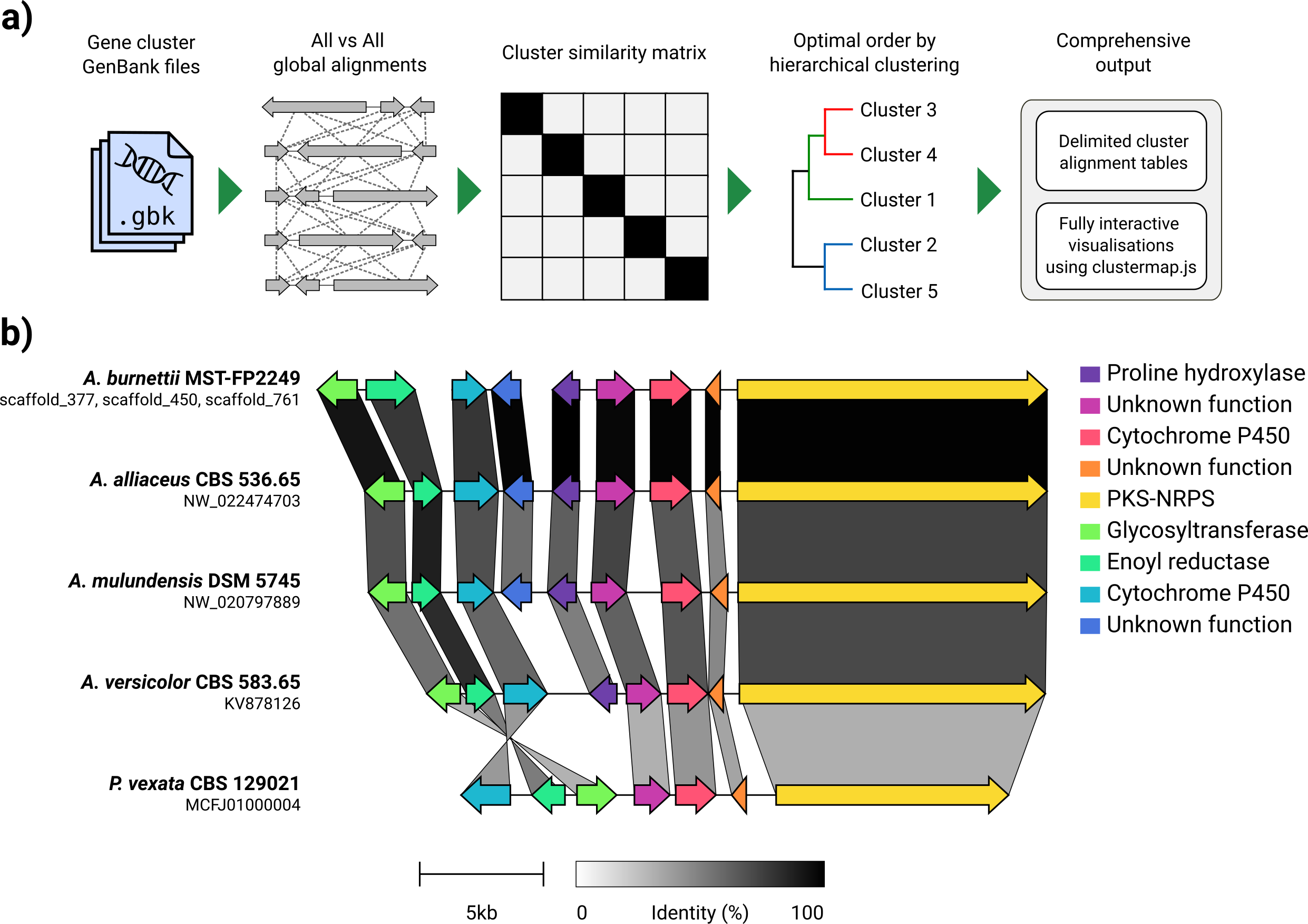
Compte tenu d'un ensemble de fichiers GenBank, Clinker extraire automatiquement les traductions protéiques, effectuera des alignements globaux entre les séquences dans chaque cluster, déterminera l'ordre d'affichage optimal basé sur la similitude du cluster et génére une visualisation interactive (en utilisant ClusterMap.js) qui peut être largement modifiée avant avant avant être exporté en tant que fichier SVG.
Clinker a été conçu principalement comme un moyen simple de visualiser des groupes de grappes de gènes biosynthétiques homologues, qui sont généralement de petites régions génomiques avec peu de gènes (comme dans l'exemple GIF). Il effectue des alignements par paire de tous les gènes dans tous les fichiers d'entrée à l'aide de l'aligneur intégré à Biopython, puis génère un document SVG interactif dans le navigateur. Le stade d'alignement évoluera très mal à plusieurs génomes avec de nombreux gènes, et la visualisation résultante sera également très lente étant donné le nombre d'éléments SVG qu'il contiendra. Si vous cherchez à aligner des génomes entiers, vous serez probablement mieux servi à l'aide d'outils construits à cette fin (par exemple, le cactus).
Clinker peut être installé directement via PIP:
pip install clinker
En clonage le code source de GitHub:
git clone https://github.com/gamcil/clinker.git
cd clinker
pip install .
Ou, par le biais de conda:
conda create -n clinker -c conda-forge -c bioconda clinker-py
conda activate clinker
Si vous avez trouvé Clinker utile, veuillez citer:
clinker & clustermap.js: Automatic generation of gene cluster comparison figures.
Gilchrist, C.L.M., Chooi, Y.-H., 2020.
Bioinformatics. doi: https://doi.org/10.1093/bioinformatics/btab007
La course à pied peut être aussi simple que:
clinker clusters/*.gbk
Cela se lira dans tous les fichiers GenBank dans le dossier, les alignera et imprimera les alignements sur le terminal. Pour générer la visualisation, utilisez l'argument -p/--plot :
clinker clusters/*.gbk -p <optional: file name to save static HTML>
Clinker peut également analyser les fichiers GFF3:
clinker cluster1.gff3 cluster2.gff3 -p
Remarque: Un fichier rapide correspondant du même nom (extensions ".fa", ".fsa", ".fna", ".fasta" ou ".faa") doit être trouvé dans le même répertoire que le GFF3, c'est-à-dire cluster1.fa et cluster2.fa .
Voir -h/--help pour plus d'informations:
usage: clinker [-h] [--version] [-r RANGES [RANGES ...]] [-gf GENE_FUNCTIONS] [-na] [-i IDENTITY] [-j JOBS] [-s SESSION] [-ji JSON_INDENT] [-f] [-o OUTPUT] [-p [PLOT]] [-dl DELIMITER] [-dc DECIMALS] [-hl] [-ha] [-mo MATRIX_OUT] [-ufo] [files ...]
clinker: Automatic creation of publication-ready gene cluster comparison figures.
clinker generates gene cluster comparison figures from GenBank files. It performs pairwise local or global alignments between every sequence in every unique pair of clusters and generates interactive, to-scale comparison figures using the clustermap.js library.
optional arguments:
-h, --help show this help message and exit
--version show program's version number and exit
Input options:
files Gene cluster GenBank files
-r RANGES [RANGES ...], --ranges RANGES [RANGES ...]
Scaffold extraction ranges. If a range is specified, only features within the range will be extracted from the scaffold. Ranges should be formatted like: scaffold:start-end (e.g. scaffold_1:15000-40000)
-gf GENE_FUNCTIONS, --gene_functions GENE_FUNCTIONS
2-column CSV file containing gene functions, used to build gene groups from same function instead of sequence similarity (e.g. GENE_001,PKS-NRPS).
Alignment options:
-na, --no_align Do not align clusters
-i IDENTITY, --identity IDENTITY
Minimum alignment sequence identity [default: 0.3]
-j JOBS, --jobs JOBS Number of alignments to run in parallel (0 to use the number of CPUs) [default: 0]
Output options:
-s SESSION, --session SESSION
Path to clinker session
-ji JSON_INDENT, --json_indent JSON_INDENT
Number of spaces to indent JSON [default: none]
-f, --force Overwrite previous output file
-o OUTPUT, --output OUTPUT
Save alignments to file
-p [PLOT], --plot [PLOT]
Plot cluster alignments using clustermap.js. If a path is given, clinker will generate a portable HTML file at that path. Otherwise, the plot will be served dynamically using Python's HTTP server.
-dl DELIMITER, --delimiter DELIMITER
Character to delimit output by [default: human readable]
-dc DECIMALS, --decimals DECIMALS
Number of decimal places in output [default: 2]
-hl, --hide_link_headers
Hide alignment column headers
-ha, --hide_aln_headers
Hide alignment cluster name headers
-mo MATRIX_OUT, --matrix_out MATRIX_OUT
Save cluster similarity matrix to file
Visualisation options:
-ufo, --use_file_order
Display clusters in order of input files
Example usage
-------------
Align clusters, plot results and print scores to screen:
$ clinker files/*.gbk
Only save gene-gene links when identity is over 50%:
$ clinker files/*.gbk -i 0.5
Save an alignment session for later:
$ clinker files/*.gbk -s session.json
Save alignments to file, in comma-delimited format, with 4 decimal places:
$ clinker files/*.gbk -o alignments.csv -dl "," -dc 4
Generate visualisation:
$ clinker files/*.gbk -p
Save visualisation as a static HTML document:
$ clinker files/*.gbk -p plot.html
Cameron Gilchrist, 2020
Par défaut, Clinker attribue automatiquement un nom et une couleur pour chaque groupe de gènes homologues. Vous pouvez plutôt pré-attribuer des noms (c.-à-d. Les fonctions) à l'aide de l'argument -gf/--gene_functions , qui prend un fichier séparé à 2 colonnes comme:
GENE_001,Cytochrome P450
GENE_002,Cytochrome P450
GENE_003,Methyltransferase
GENE_004,Methyltransferase
Cela générera deux groupes, le cytochrome P450 (GENE_001 et 002) et la méthyltransférase (Gene_003, Gene_004). Si d'autres gènes homologues sont identifiés, ils seront automatiquement ajoutés à ces groupes.
À partir de Clinker V0.0.28, vous pouvez désormais spécifier des couleurs pour les gènes définis par l'argument -gf/--gene_functions . Pour ce faire, utilisez l'argument -cm/--colour_map qui prend également un fichier CSV à 2 colonnes contenant le nom de groupe et le code couleur hexadécimal comme:
Cytochrome P450,#FF0000
Methyltransferase,#0000FF


