clinker
v0.0.30
ตอนนี้ทั้ง Cblaster และ Clinker สามารถใช้งานได้โดยไม่ต้องติดตั้งบนเว็บเซิร์ฟเวอร์ Cagecat
เครื่องกำเนิดรูปการเปรียบเทียบคลัสเตอร์ยีน
Clinker เป็นท่อสำหรับการสร้างตัวเลขการเปรียบเทียบกลุ่มยีนที่มีคุณภาพการตีพิมพ์ได้อย่างง่ายดาย
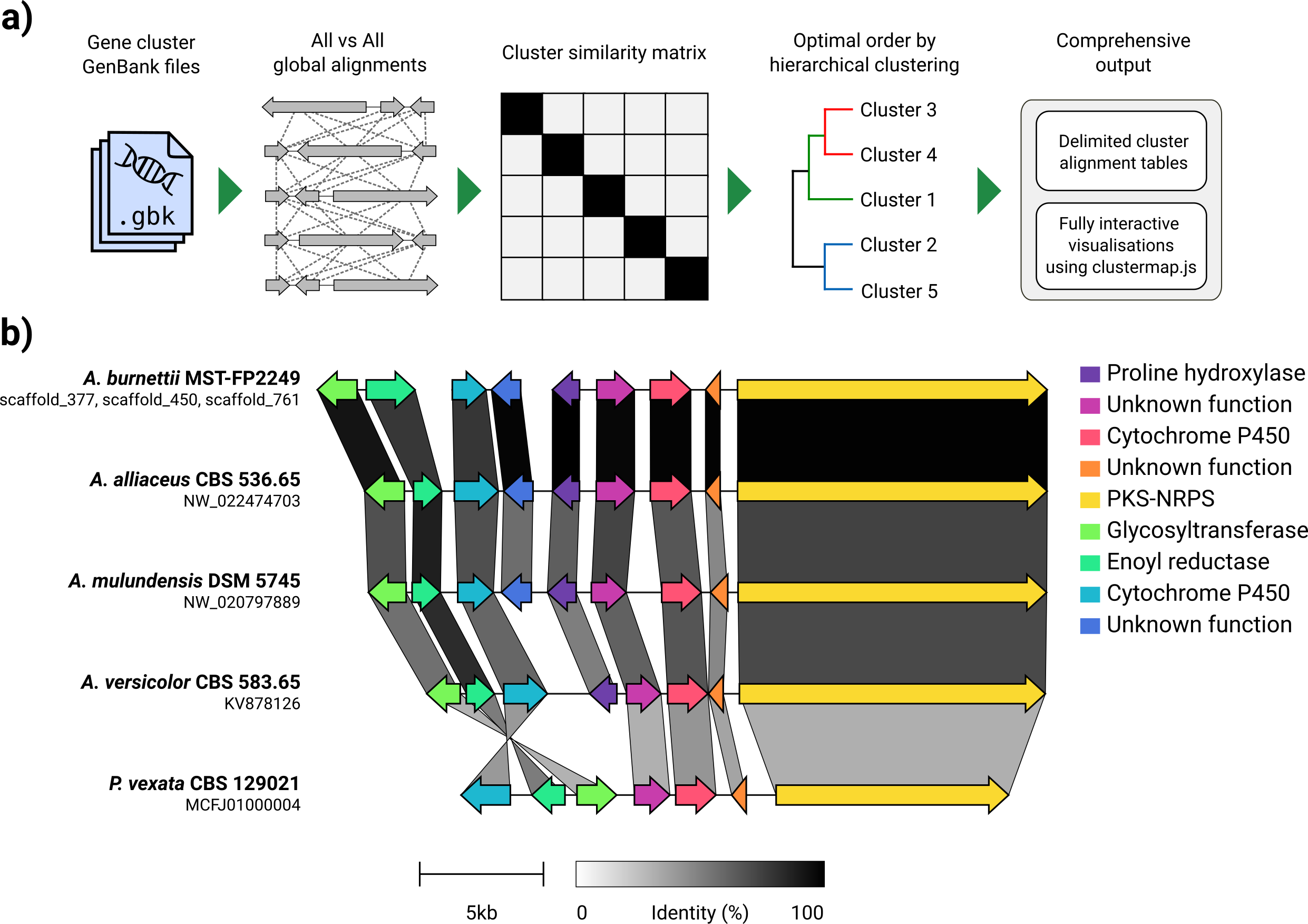
เมื่อพิจารณาจากชุดไฟล์ GenBank, Clinker จะแยกการแปลโปรตีนโดยอัตโนมัติดำเนินการจัดตำแหน่งทั่วโลกระหว่างลำดับในแต่ละคลัสเตอร์กำหนดลำดับการแสดงผลที่ดีที่สุดตามความคล้ายคลึงกันของคลัสเตอร์และสร้างการสร้างภาพข้อมูลแบบโต้ตอบ (โดยใช้ ClusterMap.js) ที่สามารถปรับแต่งได้อย่างกว้างขวางก่อน ถูกส่งออกเป็นไฟล์ SVG
Clinker ได้รับการออกแบบเป็นวิธีง่ายๆในการมองเห็นกลุ่มของกลุ่มยีนสังเคราะห์ทางชีวภาพที่คล้ายคลึงกันซึ่งโดยทั่วไปจะเป็นภูมิภาคจีโนมขนาดเล็กที่มียีนไม่มาก (เช่นในตัวอย่าง GIF) มันดำเนินการจัดตำแหน่งคู่ของยีนทั้งหมดในไฟล์อินพุตทั้งหมดโดยใช้ตัวจัดตำแหน่งที่สร้างขึ้นใน biopython จากนั้นสร้างเอกสาร SVG แบบโต้ตอบในเบราว์เซอร์ ขั้นตอนการจัดตำแหน่งจะขยายขนาดได้ไม่ดีไปจนถึงจีโนมหลายตัวที่มียีนหลายตัวและการสร้างภาพข้อมูลที่เกิดขึ้นจะช้ามากเนื่องจากมีองค์ประกอบ SVG จำนวนเท่าใดที่จะมี หากคุณกำลังมองหาการจัดตำแหน่งจีโนมทั้งหมดคุณจะได้รับการบริการที่ดีขึ้นโดยใช้เครื่องมือที่สร้างขึ้นเพื่อจุดประสงค์นั้น (เช่น Cactus)
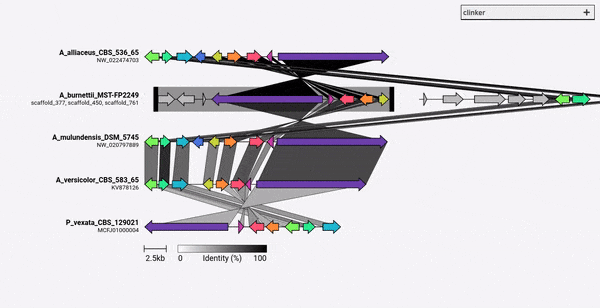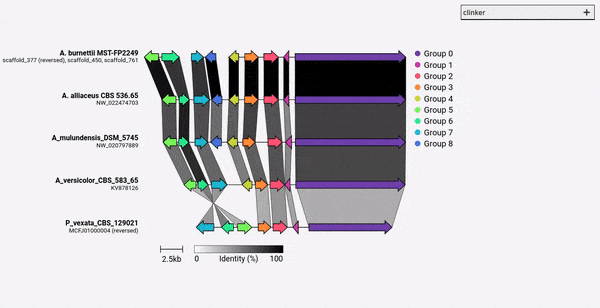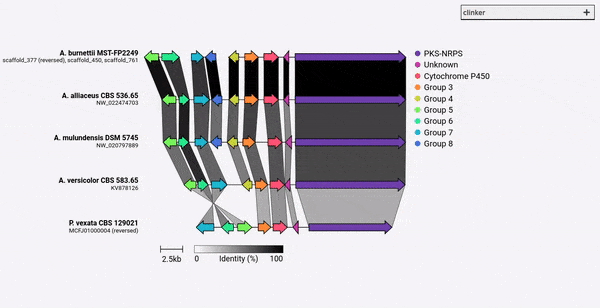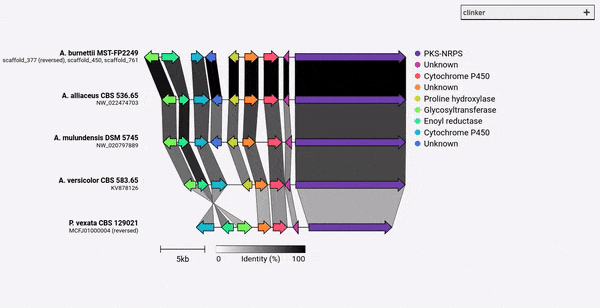
Clinker สามารถติดตั้งโดยตรงผ่าน PIP:
pip install clinker
โดยการโคลนนิ่งซอร์สโค้ดจาก GitHub:
git clone https://github.com/gamcil/clinker.git
cd clinker
pip install .
หรือผ่าน conda:
conda create -n clinker -c conda-forge -c bioconda clinker-py
conda activate clinker
หากคุณพบว่า Clinker มีประโยชน์โปรดอ้างอิง:
clinker & clustermap.js: Automatic generation of gene cluster comparison figures.
Gilchrist, C.L.M., Chooi, Y.-H., 2020.
Bioinformatics. doi: https://doi.org/10.1093/bioinformatics/btab007
การทำงานของ Clinker นั้นง่ายเหมือน:
clinker clusters/*.gbk
สิ่งนี้จะอ่านในไฟล์ GenBank ทั้งหมดภายในโฟลเดอร์จัดตำแหน่งและพิมพ์การจัดตำแหน่งไปยังเทอร์มินัล ในการสร้างการสร้างภาพให้ใช้อาร์กิวเมนต์ -p/--plot :
clinker clusters/*.gbk -p <optional: file name to save static HTML>
Clinker ยังสามารถแยกวิเคราะห์ไฟล์ GFF3:
clinker cluster1.gff3 cluster2.gff3 -p
หมายเหตุ: ไฟล์ fasta ที่สอดคล้องกันที่มีชื่อเดียวกัน (ส่วนขยาย ".fa", ".fsa", ".fna", ".fasta" หรือ ".faa") จะต้องพบในไดเรกทอรีเดียวกับ GFF3 เช่น cluster1.fa และ cluster2.fa
ดู -h/--help สำหรับข้อมูลเพิ่มเติม:
usage: clinker [-h] [--version] [-r RANGES [RANGES ...]] [-gf GENE_FUNCTIONS] [-na] [-i IDENTITY] [-j JOBS] [-s SESSION] [-ji JSON_INDENT] [-f] [-o OUTPUT] [-p [PLOT]] [-dl DELIMITER] [-dc DECIMALS] [-hl] [-ha] [-mo MATRIX_OUT] [-ufo] [files ...]
clinker: Automatic creation of publication-ready gene cluster comparison figures.
clinker generates gene cluster comparison figures from GenBank files. It performs pairwise local or global alignments between every sequence in every unique pair of clusters and generates interactive, to-scale comparison figures using the clustermap.js library.
optional arguments:
-h, --help show this help message and exit
--version show program's version number and exit
Input options:
files Gene cluster GenBank files
-r RANGES [RANGES ...], --ranges RANGES [RANGES ...]
Scaffold extraction ranges. If a range is specified, only features within the range will be extracted from the scaffold. Ranges should be formatted like: scaffold:start-end (e.g. scaffold_1:15000-40000)
-gf GENE_FUNCTIONS, --gene_functions GENE_FUNCTIONS
2-column CSV file containing gene functions, used to build gene groups from same function instead of sequence similarity (e.g. GENE_001,PKS-NRPS).
Alignment options:
-na, --no_align Do not align clusters
-i IDENTITY, --identity IDENTITY
Minimum alignment sequence identity [default: 0.3]
-j JOBS, --jobs JOBS Number of alignments to run in parallel (0 to use the number of CPUs) [default: 0]
Output options:
-s SESSION, --session SESSION
Path to clinker session
-ji JSON_INDENT, --json_indent JSON_INDENT
Number of spaces to indent JSON [default: none]
-f, --force Overwrite previous output file
-o OUTPUT, --output OUTPUT
Save alignments to file
-p [PLOT], --plot [PLOT]
Plot cluster alignments using clustermap.js. If a path is given, clinker will generate a portable HTML file at that path. Otherwise, the plot will be served dynamically using Python's HTTP server.
-dl DELIMITER, --delimiter DELIMITER
Character to delimit output by [default: human readable]
-dc DECIMALS, --decimals DECIMALS
Number of decimal places in output [default: 2]
-hl, --hide_link_headers
Hide alignment column headers
-ha, --hide_aln_headers
Hide alignment cluster name headers
-mo MATRIX_OUT, --matrix_out MATRIX_OUT
Save cluster similarity matrix to file
Visualisation options:
-ufo, --use_file_order
Display clusters in order of input files
Example usage
-------------
Align clusters, plot results and print scores to screen:
$ clinker files/*.gbk
Only save gene-gene links when identity is over 50%:
$ clinker files/*.gbk -i 0.5
Save an alignment session for later:
$ clinker files/*.gbk -s session.json
Save alignments to file, in comma-delimited format, with 4 decimal places:
$ clinker files/*.gbk -o alignments.csv -dl "," -dc 4
Generate visualisation:
$ clinker files/*.gbk -p
Save visualisation as a static HTML document:
$ clinker files/*.gbk -p plot.html
Cameron Gilchrist, 2020
โดยค่าเริ่มต้น Clinker จะกำหนดชื่อและสีสำหรับแต่ละกลุ่มของยีน homologous โดยอัตโนมัติ คุณสามารถแทนที่ชื่อล่วงหน้า (เช่นฟังก์ชั่น) โดยใช้อาร์กิวเมนต์ -gf/--gene_functions ซึ่งใช้ไฟล์ที่คั่นด้วยเครื่องหมายจุลภาค 2 คอลัมน์เช่น:
GENE_001,Cytochrome P450
GENE_002,Cytochrome P450
GENE_003,Methyltransferase
GENE_004,Methyltransferase
สิ่งนี้จะสร้างสองกลุ่มคือ Cytochrome P450 (Gene_001 และ 002) และ methyltransferase (Gene_003, Gene_004) หากมีการระบุยีนที่คล้ายคลึงกันอื่น ๆ พวกเขาจะถูกเพิ่มเข้าไปในกลุ่มเหล่านี้โดยอัตโนมัติ
ในฐานะของ Clinker v0.0.28 ตอนนี้คุณสามารถระบุสีสำหรับยีนที่กำหนดโดยอาร์กิวเมนต์ -gf/--gene_functions ในการทำเช่นนี้ให้ใช้อาร์กิวเมนต์ -cm/--colour_map ซึ่งใช้ไฟล์ CSV 2 คอลัมน์ที่มีชื่อกลุ่มและรหัสสีเลขฐานสิบหกเช่น:
Cytochrome P450,#FF0000
Methyltransferase,#0000FF


