clinker
v0.0.30
O CBLASTER e o Clinker agora podem ser usados sem instalação no servidor da web Cagecat.
Gerador de figuras de comparação de cluster de genes
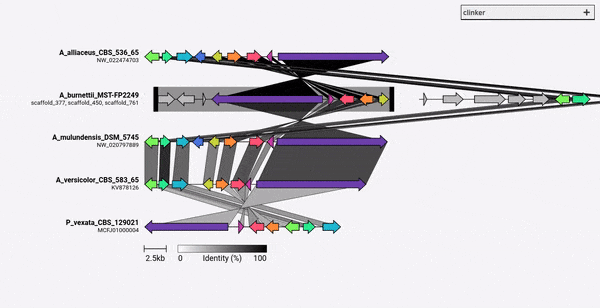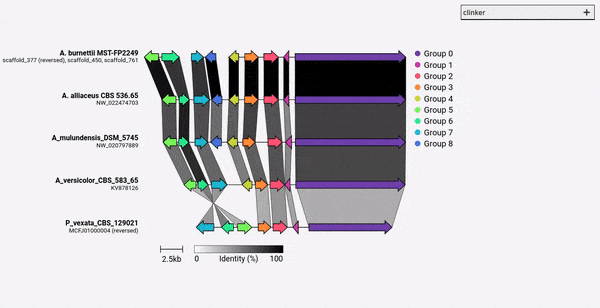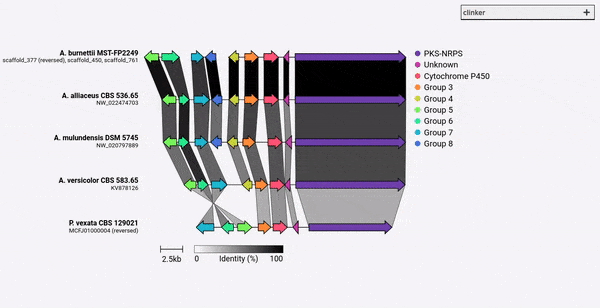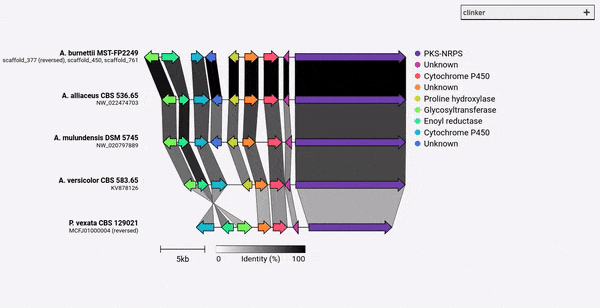
Clinker é um pipeline para gerar facilmente as figuras de comparação de cluster de qualidade de qualidade de publicação.
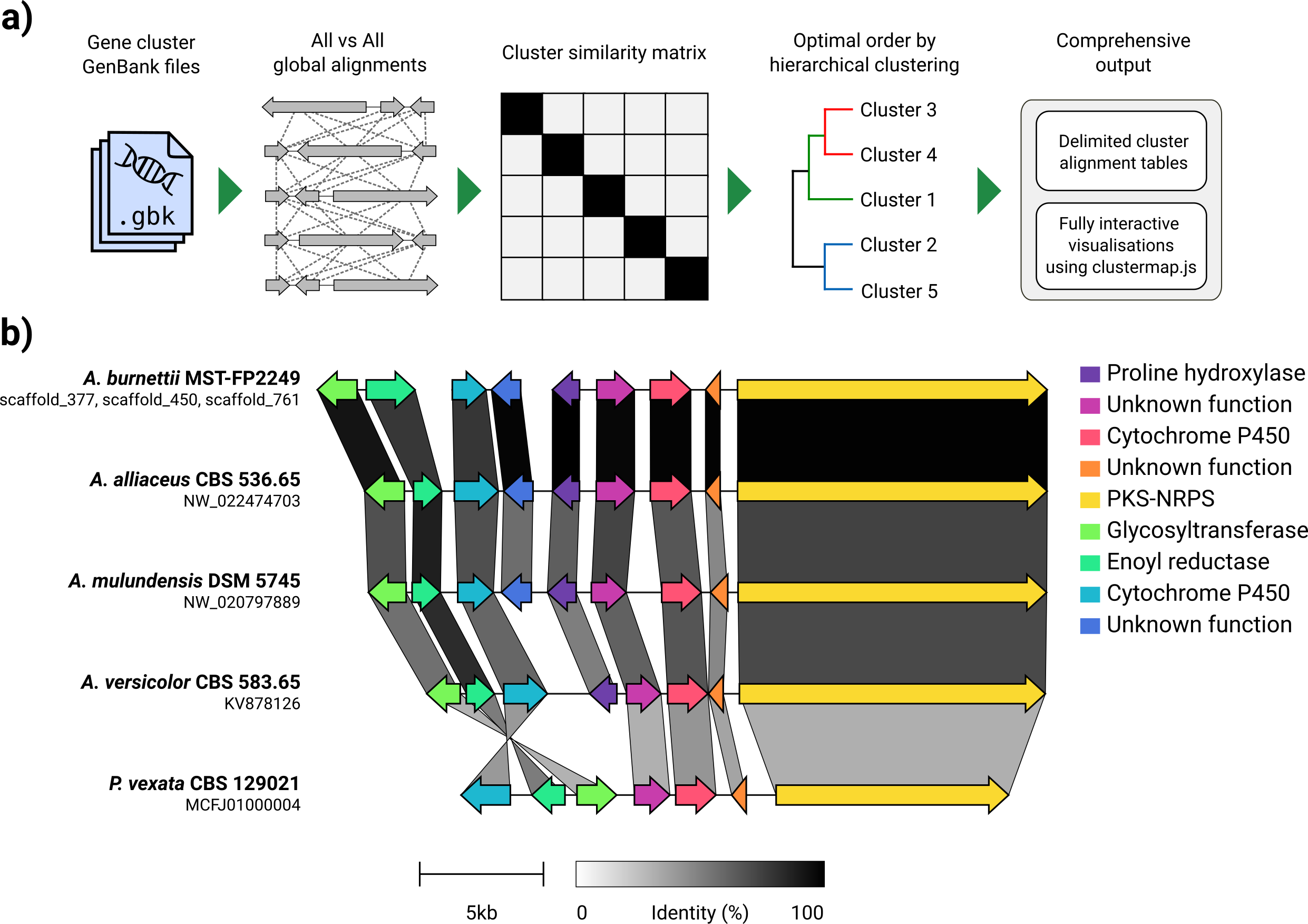
Dado um conjunto de arquivos GenBank, o Clinker extrairá automaticamente as traduções de proteínas, realizará alinhamentos globais entre sequências em cada cluster, determinará a ordem de exibição ideal com base na similaridade do cluster e gerar uma visualização interativa (usando clustermap.js) que pode ser extensivamente ajustada antes sendo exportado como um arquivo SVG.
Clinker foi projetado principalmente como uma maneira simples de visualizar grupos de aglomerados de genes biossintéticos homólogos, que são tipicamente pequenas regiões genômicas com poucos genes (como no exemplo gif). Ele executa alinhamentos em pares de todos os genes em todos os arquivos de entrada usando o alinhador embutido no Biopython e gera um documento SVG interativo no navegador. O estágio de alinhamento será muito ruim para múltiplos genomas com muitos genes, e a visualização resultante também será muito lenta, considerando quantos elementos SVG conterão. Se você deseja alinhar genomas inteiros, provavelmente será melhor servido usando ferramentas construídas para esse fim (por exemplo, cacto).
Clinker pode ser instalado diretamente através do PIP:
pip install clinker
Clonando o código -fonte do GitHub:
git clone https://github.com/gamcil/clinker.git
cd clinker
pip install .
Ou, através de conda:
conda create -n clinker -c conda-forge -c bioconda clinker-py
conda activate clinker
Se você achou o Clinker útil, cite:
clinker & clustermap.js: Automatic generation of gene cluster comparison figures.
Gilchrist, C.L.M., Chooi, Y.-H., 2020.
Bioinformatics. doi: https://doi.org/10.1093/bioinformatics/btab007
O clinker em execução pode ser tão simples quanto:
clinker clusters/*.gbk
Isso lerá em todos os arquivos GenBank dentro da pasta, alinham -os e imprimirão os alinhamentos ao terminal. Para gerar a visualização, use o argumento -p/--plot :
clinker clusters/*.gbk -p <optional: file name to save static HTML>
O Clinker também pode analisar os arquivos GFF3:
clinker cluster1.gff3 cluster2.gff3 -p
Nota: Um arquivo fasta correspondente com o mesmo nome (extensões ".fa", ".fsa", ".fna", ".fastA" ou ".faa") deve ser encontrado no mesmo diretório que o GFF3, ou seja, cluster1.fa e cluster2.fa .
Veja -h/--help para obter mais informações:
usage: clinker [-h] [--version] [-r RANGES [RANGES ...]] [-gf GENE_FUNCTIONS] [-na] [-i IDENTITY] [-j JOBS] [-s SESSION] [-ji JSON_INDENT] [-f] [-o OUTPUT] [-p [PLOT]] [-dl DELIMITER] [-dc DECIMALS] [-hl] [-ha] [-mo MATRIX_OUT] [-ufo] [files ...]
clinker: Automatic creation of publication-ready gene cluster comparison figures.
clinker generates gene cluster comparison figures from GenBank files. It performs pairwise local or global alignments between every sequence in every unique pair of clusters and generates interactive, to-scale comparison figures using the clustermap.js library.
optional arguments:
-h, --help show this help message and exit
--version show program's version number and exit
Input options:
files Gene cluster GenBank files
-r RANGES [RANGES ...], --ranges RANGES [RANGES ...]
Scaffold extraction ranges. If a range is specified, only features within the range will be extracted from the scaffold. Ranges should be formatted like: scaffold:start-end (e.g. scaffold_1:15000-40000)
-gf GENE_FUNCTIONS, --gene_functions GENE_FUNCTIONS
2-column CSV file containing gene functions, used to build gene groups from same function instead of sequence similarity (e.g. GENE_001,PKS-NRPS).
Alignment options:
-na, --no_align Do not align clusters
-i IDENTITY, --identity IDENTITY
Minimum alignment sequence identity [default: 0.3]
-j JOBS, --jobs JOBS Number of alignments to run in parallel (0 to use the number of CPUs) [default: 0]
Output options:
-s SESSION, --session SESSION
Path to clinker session
-ji JSON_INDENT, --json_indent JSON_INDENT
Number of spaces to indent JSON [default: none]
-f, --force Overwrite previous output file
-o OUTPUT, --output OUTPUT
Save alignments to file
-p [PLOT], --plot [PLOT]
Plot cluster alignments using clustermap.js. If a path is given, clinker will generate a portable HTML file at that path. Otherwise, the plot will be served dynamically using Python's HTTP server.
-dl DELIMITER, --delimiter DELIMITER
Character to delimit output by [default: human readable]
-dc DECIMALS, --decimals DECIMALS
Number of decimal places in output [default: 2]
-hl, --hide_link_headers
Hide alignment column headers
-ha, --hide_aln_headers
Hide alignment cluster name headers
-mo MATRIX_OUT, --matrix_out MATRIX_OUT
Save cluster similarity matrix to file
Visualisation options:
-ufo, --use_file_order
Display clusters in order of input files
Example usage
-------------
Align clusters, plot results and print scores to screen:
$ clinker files/*.gbk
Only save gene-gene links when identity is over 50%:
$ clinker files/*.gbk -i 0.5
Save an alignment session for later:
$ clinker files/*.gbk -s session.json
Save alignments to file, in comma-delimited format, with 4 decimal places:
$ clinker files/*.gbk -o alignments.csv -dl "," -dc 4
Generate visualisation:
$ clinker files/*.gbk -p
Save visualisation as a static HTML document:
$ clinker files/*.gbk -p plot.html
Cameron Gilchrist, 2020
Por padrão, o Clinker atribui automaticamente um nome e cor para cada grupo de genes homólogos. Em vez disso, você pode pré-assinar nomes (ou seja, funções) usando o argumento -gf/--gene_functions , que leva um arquivo separado por vírgula de duas colunas como:
GENE_001,Cytochrome P450
GENE_002,Cytochrome P450
GENE_003,Methyltransferase
GENE_004,Methyltransferase
Isso gerará dois grupos, o citocromo p450 (gene_001 e 002) e a metiltransferase (gene_003, gene_004). Se houver outros genes homólogos, eles serão adicionados automaticamente a esses grupos.
A partir do clinker v0.0.28, agora você pode especificar cores para genes definidos pelo argumento -gf/--gene_functions . Para fazer isso, use o argumento -cm/--colour_map , que também leva um arquivo CSV de 2 colunas que contém o nome do grupo e o código de cores hexadecimal como:
Cytochrome P450,#FF0000
Methyltransferase,#0000FF


