clinker
v0.0.30
Как Cblaster, так и клинкер теперь могут использоваться без установки на CageCat Webserver.
Сравнение генов Генератор рисунок.
Clinker-это трубопровод для легко генерирующих цифр сравнения генов качества публикации.
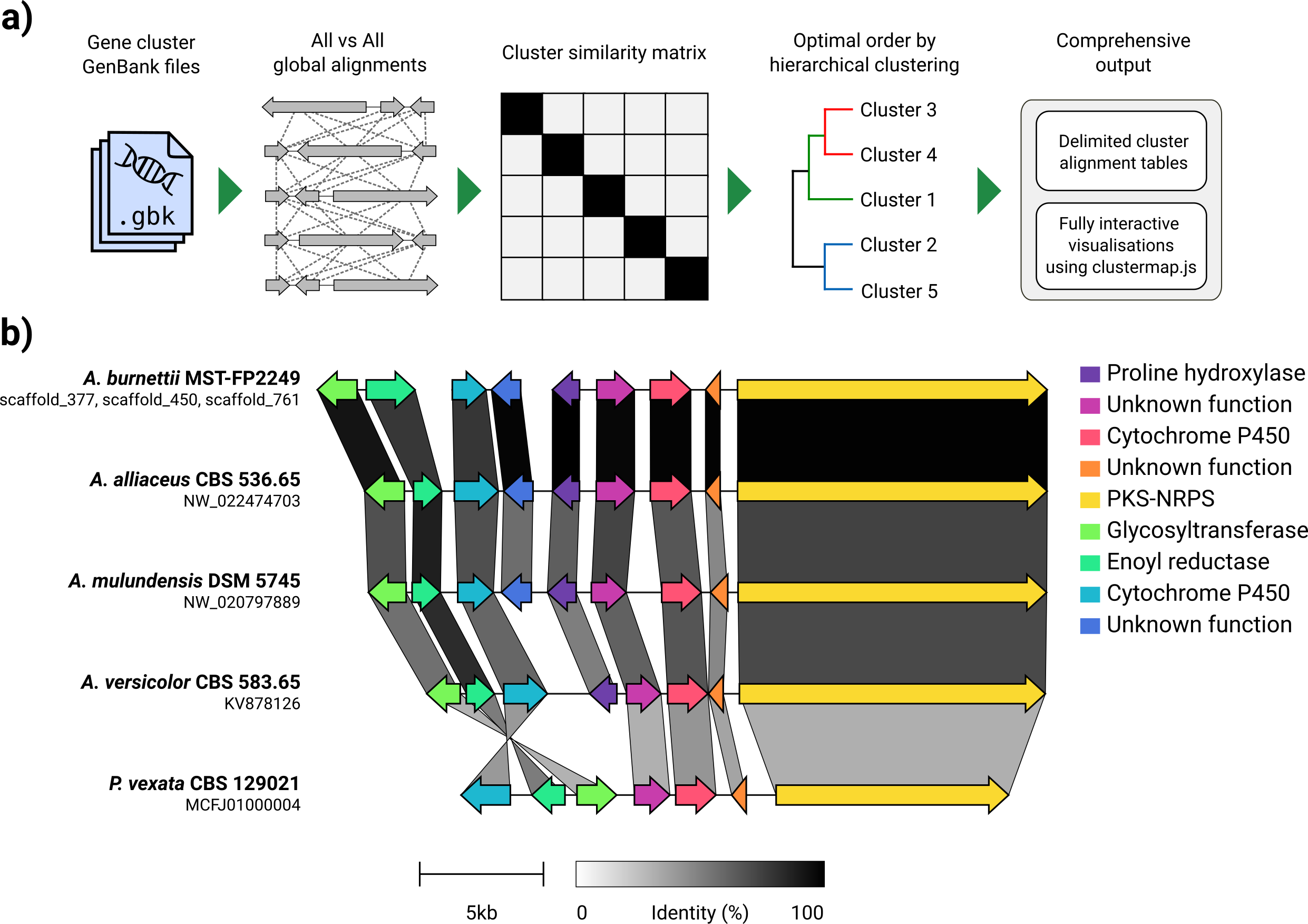
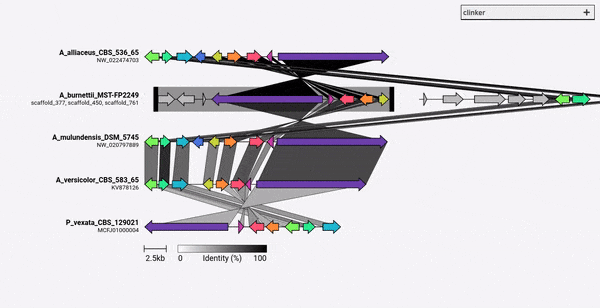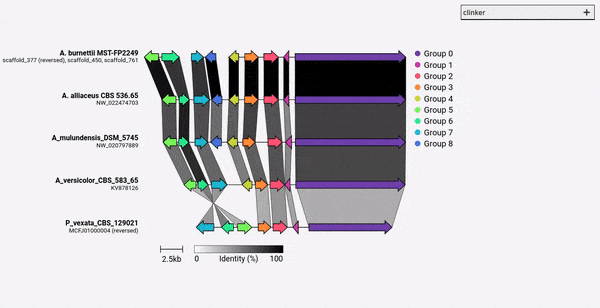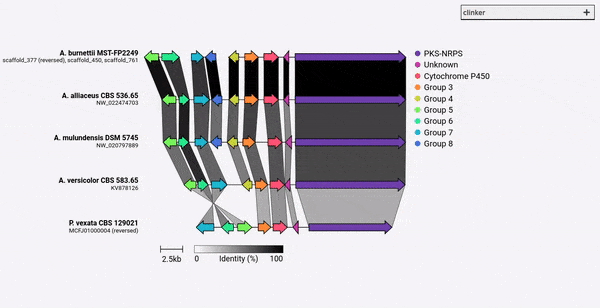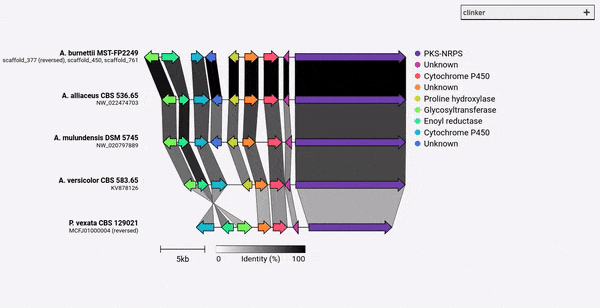
Учитывая набор файлов GenBank, клинкер автоматически извлечет трансляции белка, выполняет глобальные выравнивания между последовательностями в каждом кластере, определяет оптимальный порядок отображения на основе сходства кластера и генерировать интерактивную визуализацию (с использованием clustermap.js), который может быть широко настроен ранее экспортируется как файл SVG.
Клинкер был разработан главным образом как простой способ визуализации групп гомологичных генов гомологичных биосинтетических генов, которые обычно представляют собой небольшие геномные области с не большим количеством генов (как в примере GIF). Он выполняет парные выравнивания всех генов во всех входных файлах, используя Aligner, встроенный в BiopyThon, затем генерирует интерактивный документ SVG в браузере. Стадия выравнивания будет очень плохо масштабироваться до нескольких геномов со многими генами, и полученная визуализация также будет очень медленной, учитывая, сколько элементов SVG он будет содержать. Если вы хотите выравнивать целые геномы, вы, вероятно, будете лучше обслуживаться, используя инструменты, созданные для этой цели (например, кактус).
Клинкер может быть установлен непосредственно через PIP:
pip install clinker
Клонируя исходный код от GitHub:
git clone https://github.com/gamcil/clinker.git
cd clinker
pip install .
Или через Conda:
conda create -n clinker -c conda-forge -c bioconda clinker-py
conda activate clinker
Если вы нашли Clinker полезным, пожалуйста, цитируйте:
clinker & clustermap.js: Automatic generation of gene cluster comparison figures.
Gilchrist, C.L.M., Chooi, Y.-H., 2020.
Bioinformatics. doi: https://doi.org/10.1093/bioinformatics/btab007
Запуск клинкера может быть таким же простым, как:
clinker clusters/*.gbk
Это будет читать во всех файлах GenBank в папке, выравнивает их и распечатает выравнивания на терминал. Чтобы генерировать визуализацию, используйте аргумент -p/--plot :
clinker clusters/*.gbk -p <optional: file name to save static HTML>
Clinker также может анализировать файлы GFF3:
clinker cluster1.gff3 cluster2.gff3 -p
Примечание. Соответствующий файл FASTA с тем же именем (расширения ".fa", ".fsa", ".fna", ".fasta" или ".faa") должен быть найден в том же каталоге, что и GFF3, то есть cluster1.fa и cluster2.fa .
Смотрите -h/--help для получения дополнительной информации:
usage: clinker [-h] [--version] [-r RANGES [RANGES ...]] [-gf GENE_FUNCTIONS] [-na] [-i IDENTITY] [-j JOBS] [-s SESSION] [-ji JSON_INDENT] [-f] [-o OUTPUT] [-p [PLOT]] [-dl DELIMITER] [-dc DECIMALS] [-hl] [-ha] [-mo MATRIX_OUT] [-ufo] [files ...]
clinker: Automatic creation of publication-ready gene cluster comparison figures.
clinker generates gene cluster comparison figures from GenBank files. It performs pairwise local or global alignments between every sequence in every unique pair of clusters and generates interactive, to-scale comparison figures using the clustermap.js library.
optional arguments:
-h, --help show this help message and exit
--version show program's version number and exit
Input options:
files Gene cluster GenBank files
-r RANGES [RANGES ...], --ranges RANGES [RANGES ...]
Scaffold extraction ranges. If a range is specified, only features within the range will be extracted from the scaffold. Ranges should be formatted like: scaffold:start-end (e.g. scaffold_1:15000-40000)
-gf GENE_FUNCTIONS, --gene_functions GENE_FUNCTIONS
2-column CSV file containing gene functions, used to build gene groups from same function instead of sequence similarity (e.g. GENE_001,PKS-NRPS).
Alignment options:
-na, --no_align Do not align clusters
-i IDENTITY, --identity IDENTITY
Minimum alignment sequence identity [default: 0.3]
-j JOBS, --jobs JOBS Number of alignments to run in parallel (0 to use the number of CPUs) [default: 0]
Output options:
-s SESSION, --session SESSION
Path to clinker session
-ji JSON_INDENT, --json_indent JSON_INDENT
Number of spaces to indent JSON [default: none]
-f, --force Overwrite previous output file
-o OUTPUT, --output OUTPUT
Save alignments to file
-p [PLOT], --plot [PLOT]
Plot cluster alignments using clustermap.js. If a path is given, clinker will generate a portable HTML file at that path. Otherwise, the plot will be served dynamically using Python's HTTP server.
-dl DELIMITER, --delimiter DELIMITER
Character to delimit output by [default: human readable]
-dc DECIMALS, --decimals DECIMALS
Number of decimal places in output [default: 2]
-hl, --hide_link_headers
Hide alignment column headers
-ha, --hide_aln_headers
Hide alignment cluster name headers
-mo MATRIX_OUT, --matrix_out MATRIX_OUT
Save cluster similarity matrix to file
Visualisation options:
-ufo, --use_file_order
Display clusters in order of input files
Example usage
-------------
Align clusters, plot results and print scores to screen:
$ clinker files/*.gbk
Only save gene-gene links when identity is over 50%:
$ clinker files/*.gbk -i 0.5
Save an alignment session for later:
$ clinker files/*.gbk -s session.json
Save alignments to file, in comma-delimited format, with 4 decimal places:
$ clinker files/*.gbk -o alignments.csv -dl "," -dc 4
Generate visualisation:
$ clinker files/*.gbk -p
Save visualisation as a static HTML document:
$ clinker files/*.gbk -p plot.html
Cameron Gilchrist, 2020
По умолчанию Clinker автоматически назначает имя и цвет для каждой группы гомологичных генов. Вместо этого вы можете предварительно согласовать имена (т.е. функции), используя аргумент -gf/--gene_functions , который принимает файл, разделенный с запятыми с двумя колонками, подобен:
GENE_001,Cytochrome P450
GENE_002,Cytochrome P450
GENE_003,Methyltransferase
GENE_004,Methyltransferase
Это генерирует две группы: цитохром P450 (Gene_001 и 002) и метилтрансфераза (Gene_003, Gene_004). Если есть какие -либо другие гомологичные гены, они автоматически будут добавлены в эти группы.
По состоянию на Clinker v0.0.28, теперь вы можете указать цвета для генов, определенных аргументом -gf/--gene_functions . Для этого используйте аргумент -cm/--colour_map , который также принимает 2-колодский файл CSV, содержащий имя группы и шестнадцатеричный цветовой код, например:
Cytochrome P450,#FF0000
Methyltransferase,#0000FF


