clinker
v0.0.30
CblasterとClinkerの両方が、Cagecat Webサーバーに設置せずに使用できるようになりました。
遺伝子クラスター比較図ジェネレーター
Clinkerは、出版物質の遺伝子クラスター比較図を簡単に生成するためのパイプラインです。
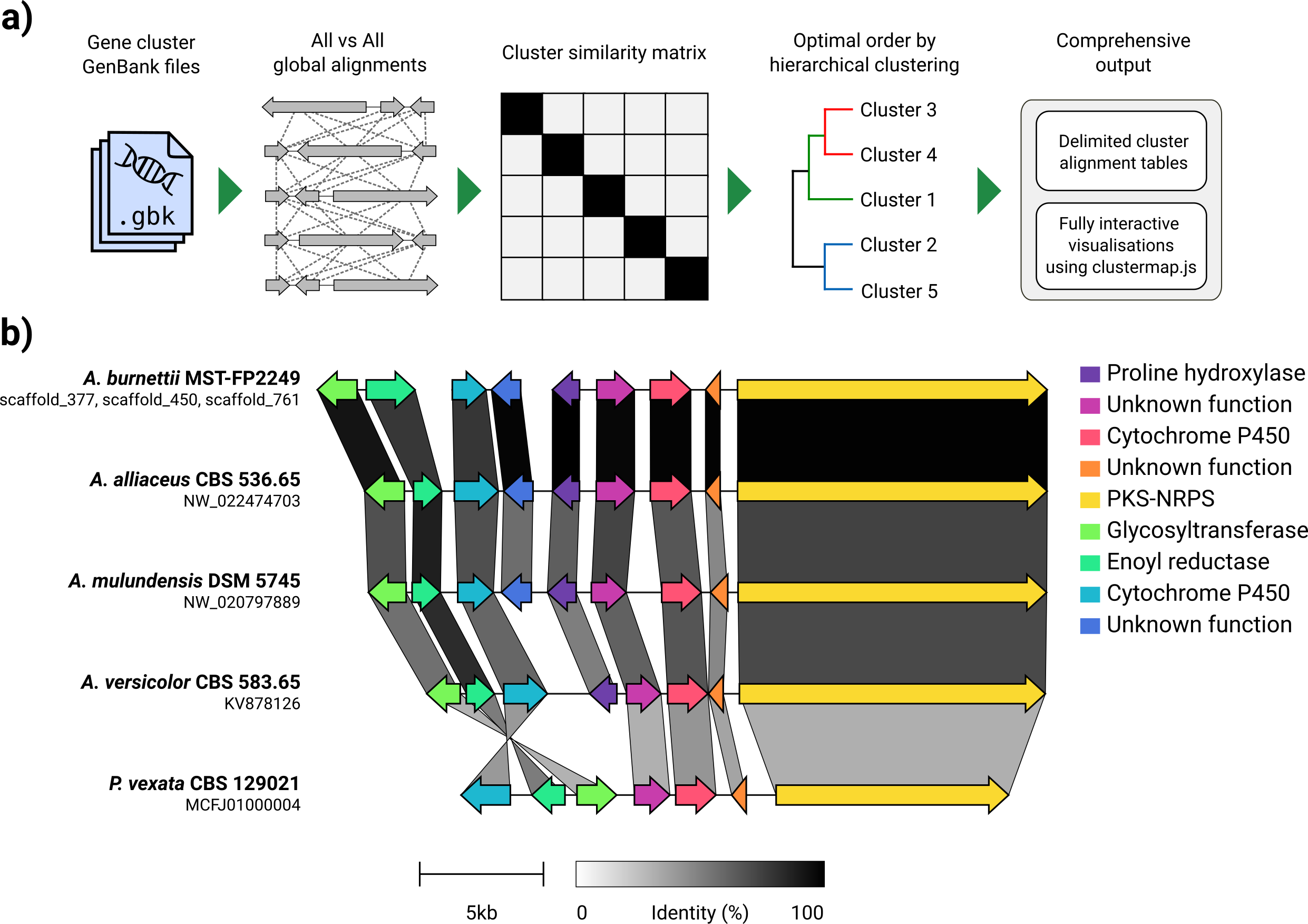
GenBankファイルのセットを考慮して、Clinkerはタンパク質翻訳を自動的に抽出し、各クラスター内のシーケンス間でグローバルなアライメントを実行し、クラスターの類似性に基づいて最適な表示順序を決定し、以前に広範囲に調整できるインタラクティブな視覚化(ClusterMap.jsを使用)を生成します。 SVGファイルとしてエクスポートされています。
Clinkerは、主に相同生的な合成遺伝子クラスターのグループを視覚化する簡単な方法として設計されました。これは、通常、遺伝子があまりない小さなゲノム領域です(例のように)。 BioPythonに組み込まれたAlignerを使用して、すべての入力ファイルのすべての遺伝子のペアワイズアラインメントを実行し、ブラウザーでインタラクティブSVGドキュメントを生成します。アライメント段階は、多くの遺伝子を使用した複数のゲノムに対して非常に不十分にスケーリングされ、結果として生じる視覚化は、SVG要素の数が含まれることを考えると非常に遅くなります。ゲノム全体を調整したい場合は、その目的のために構築されたツール(サボテンなど)を使用して、より良いサービスを提供する可能性があります。
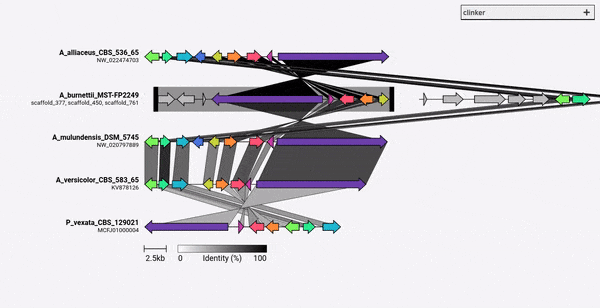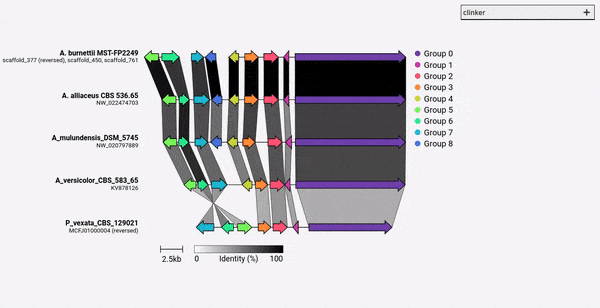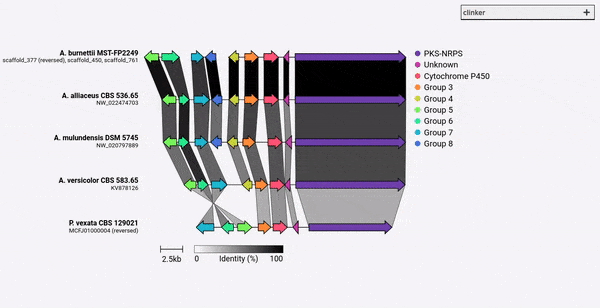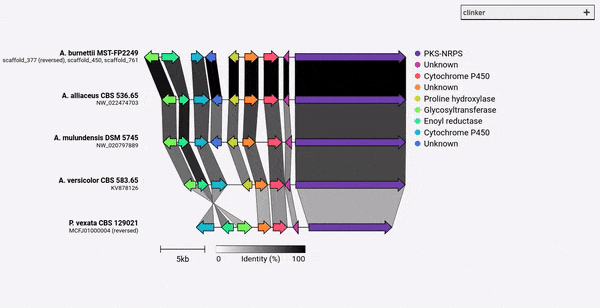
クリンカーは、PIPを介して直接インストールできます。
pip install clinker
GitHubからソースコードをクローニングすることにより:
git clone https://github.com/gamcil/clinker.git
cd clinker
pip install .
または、コンドラを介して:
conda create -n clinker -c conda-forge -c bioconda clinker-py
conda activate clinker
Clinkerが便利だと思ったら、引用してください:
clinker & clustermap.js: Automatic generation of gene cluster comparison figures.
Gilchrist, C.L.M., Chooi, Y.-H., 2020.
Bioinformatics. doi: https://doi.org/10.1093/bioinformatics/btab007
クリンカーを実行することは、次のように簡単になります。
clinker clusters/*.gbk
これにより、フォルダー内のすべてのGenBankファイルで読み取り、それらを調整し、ターミナルのアライメントを印刷します。視覚化を生成するには、 -p/--plot引数を使用します。
clinker clusters/*.gbk -p <optional: file name to save static HTML>
ClinkerはGFF3ファイルを解析することもできます。
clinker cluster1.gff3 cluster2.gff3 -p
注:同じ名前(拡張子 ".fa"、 ".fsa"、 ".fna"、 ".fasta"または ".faa")の対応するFASTAファイルは、GFF3、IE cluster1.faと同じディレクトリにある必要があります。 cluster1.faおよびcluster2.fa 。
詳細については-h/--helpを参照してください。
usage: clinker [-h] [--version] [-r RANGES [RANGES ...]] [-gf GENE_FUNCTIONS] [-na] [-i IDENTITY] [-j JOBS] [-s SESSION] [-ji JSON_INDENT] [-f] [-o OUTPUT] [-p [PLOT]] [-dl DELIMITER] [-dc DECIMALS] [-hl] [-ha] [-mo MATRIX_OUT] [-ufo] [files ...]
clinker: Automatic creation of publication-ready gene cluster comparison figures.
clinker generates gene cluster comparison figures from GenBank files. It performs pairwise local or global alignments between every sequence in every unique pair of clusters and generates interactive, to-scale comparison figures using the clustermap.js library.
optional arguments:
-h, --help show this help message and exit
--version show program's version number and exit
Input options:
files Gene cluster GenBank files
-r RANGES [RANGES ...], --ranges RANGES [RANGES ...]
Scaffold extraction ranges. If a range is specified, only features within the range will be extracted from the scaffold. Ranges should be formatted like: scaffold:start-end (e.g. scaffold_1:15000-40000)
-gf GENE_FUNCTIONS, --gene_functions GENE_FUNCTIONS
2-column CSV file containing gene functions, used to build gene groups from same function instead of sequence similarity (e.g. GENE_001,PKS-NRPS).
Alignment options:
-na, --no_align Do not align clusters
-i IDENTITY, --identity IDENTITY
Minimum alignment sequence identity [default: 0.3]
-j JOBS, --jobs JOBS Number of alignments to run in parallel (0 to use the number of CPUs) [default: 0]
Output options:
-s SESSION, --session SESSION
Path to clinker session
-ji JSON_INDENT, --json_indent JSON_INDENT
Number of spaces to indent JSON [default: none]
-f, --force Overwrite previous output file
-o OUTPUT, --output OUTPUT
Save alignments to file
-p [PLOT], --plot [PLOT]
Plot cluster alignments using clustermap.js. If a path is given, clinker will generate a portable HTML file at that path. Otherwise, the plot will be served dynamically using Python's HTTP server.
-dl DELIMITER, --delimiter DELIMITER
Character to delimit output by [default: human readable]
-dc DECIMALS, --decimals DECIMALS
Number of decimal places in output [default: 2]
-hl, --hide_link_headers
Hide alignment column headers
-ha, --hide_aln_headers
Hide alignment cluster name headers
-mo MATRIX_OUT, --matrix_out MATRIX_OUT
Save cluster similarity matrix to file
Visualisation options:
-ufo, --use_file_order
Display clusters in order of input files
Example usage
-------------
Align clusters, plot results and print scores to screen:
$ clinker files/*.gbk
Only save gene-gene links when identity is over 50%:
$ clinker files/*.gbk -i 0.5
Save an alignment session for later:
$ clinker files/*.gbk -s session.json
Save alignments to file, in comma-delimited format, with 4 decimal places:
$ clinker files/*.gbk -o alignments.csv -dl "," -dc 4
Generate visualisation:
$ clinker files/*.gbk -p
Save visualisation as a static HTML document:
$ clinker files/*.gbk -p plot.html
Cameron Gilchrist, 2020
デフォルトでは、Clinkerは相同遺伝子の各グループに名前と色を自動的に割り当てます。代わりに、 -gf/--gene_functions引数を使用して名前(すなわち関数)を割り当てることができます。
GENE_001,Cytochrome P450
GENE_002,Cytochrome P450
GENE_003,Methyltransferase
GENE_004,Methyltransferase
これにより、シトクロムP450(GENE_001および002)とメチルトランスフェラーゼ(GENE_003、GENE_004)の2つのグループが生成されます。他の相同遺伝子が特定されている場合、それらはこれらのグループに自動的に追加されます。
Clinker v0.0.28の時点で、 -gf/--gene_functions引数によって定義された遺伝子の色を指定できるようになりました。これを行うには、グループ名と16進数のカラーコードを含む2列のCSVファイルも取得する-cm/--colour_map引数を使用します。
Cytochrome P450,#FF0000
Methyltransferase,#0000FF


