clinker
v0.0.30
Sowohl Cblaster als auch Clinker können jetzt ohne Installation auf dem Cagecat -Webserver verwendet werden.
Gen -Cluster -Vergleichsgenerator
CLINKER ist eine Pipeline für die einfache Erzeugung von Gen-Cluster-Vergleichsfiguren in Publikationsqualität.
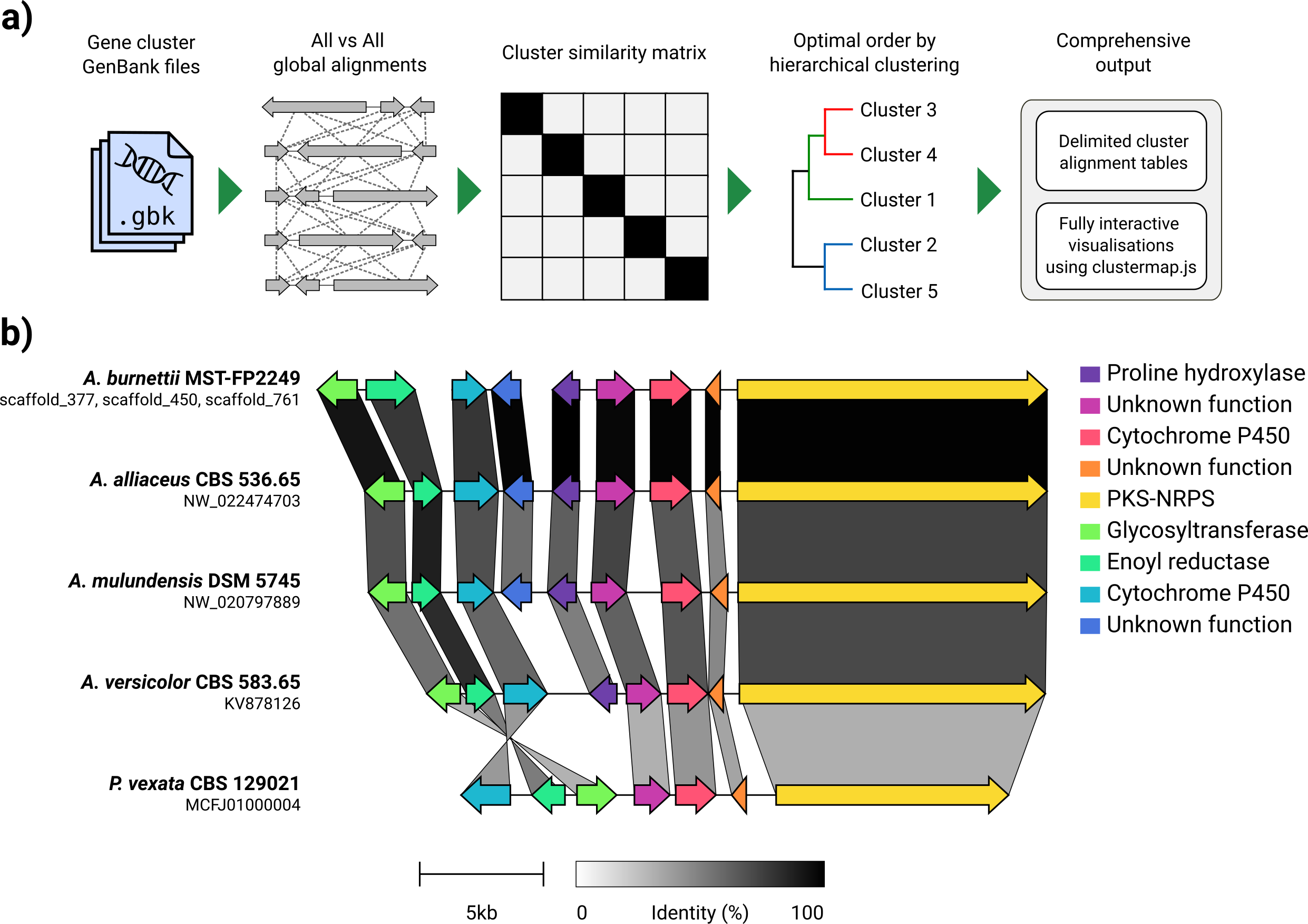
Bei einem Satz von GenBank -Dateien extrahiert Clinker automatisch Proteintranslationen, führt globale Ausrichtungen zwischen den Sequenzen in jedem Cluster durch, bestimmen die optimale Anzeigereihenfolge basierend auf der Ähnlichkeit von Cluster und generieren eine interaktive Visualisierung (mit Clustermap.js), die zuvor umfassend optimiert werden kann als SVG -Datei exportiert werden.
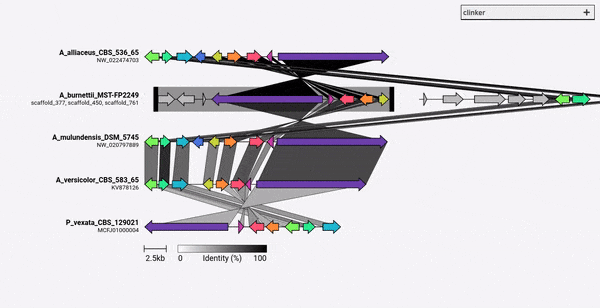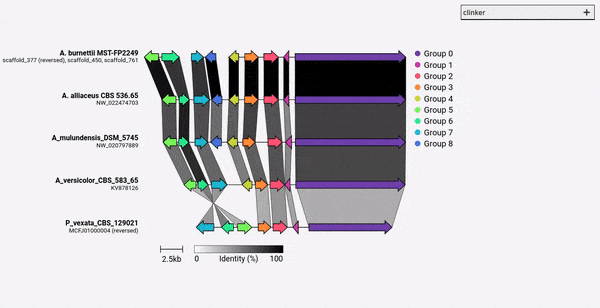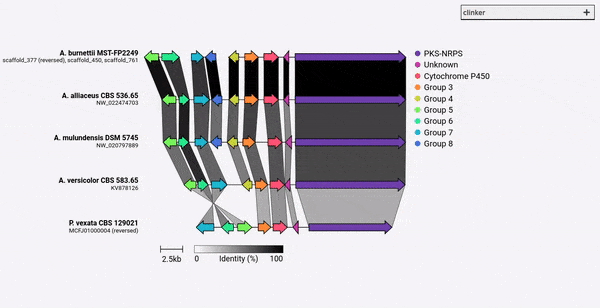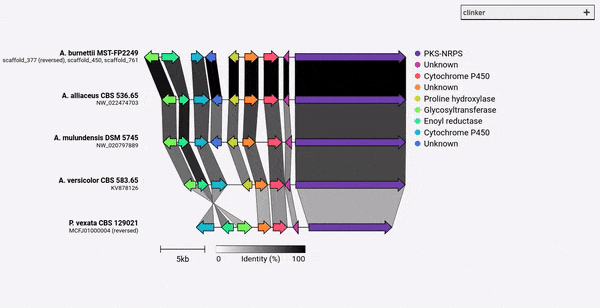
Clinker wurde hauptsächlich als einfache Möglichkeit entwickelt, Gruppen homologer biosynthetischer Gencluster zu visualisieren, die typischerweise kleine genomische Regionen mit nicht vielen Genen sind (wie im Beispiel GIF). Es führt paarweise Alignments aller Gene in allen Eingabedateien mit dem in Biopython integrierten Aligner durch und erzeugt dann ein interaktives SVG -Dokument im Browser. Die Ausrichtungsphase wird mit vielen Genen sehr schlecht auf mehrere Genome skalieren, und die daraus resultierende Visualisierung wird auch sehr langsam sein, da sie viele SVG -Elemente enthalten werden. Wenn Sie alle Genome ausrichten möchten, werden Sie wahrscheinlich besser mit Tools bedient, die für diesen Zweck erstellt wurden (z. B. Kaktus).
Clinker kann direkt über PIP installiert werden:
pip install clinker
Durch Klonen des Quellcode aus GitHub:
git clone https://github.com/gamcil/clinker.git
cd clinker
pip install .
Oder durch Conda:
conda create -n clinker -c conda-forge -c bioconda clinker-py
conda activate clinker
Wenn Sie Clinker nützlich empfanden, zitieren Sie bitte:
clinker & clustermap.js: Automatic generation of gene cluster comparison figures.
Gilchrist, C.L.M., Chooi, Y.-H., 2020.
Bioinformatics. doi: https://doi.org/10.1093/bioinformatics/btab007
Das Ausführen von Klinker kann so einfach sein wie:
clinker clusters/*.gbk
Dies wird in allen GenBank -Dateien im Ordner gelesen, sie ausgerichtet und die Ausrichtungen auf das Terminal drucken. Um die Visualisierung zu erzeugen, verwenden Sie das Argument -p/--plot :
clinker clusters/*.gbk -p <optional: file name to save static HTML>
Clinker kann auch GFF3 -Dateien analysieren:
clinker cluster1.gff3 cluster2.gff3 -p
Hinweis: Eine entsprechende Fasta -Datei gleichername (Erweiterungen ".fa", ".fsa", ".fna", ".fasta" oder ".faa") muss im selben Verzeichnis wie das GFF3 gefunden werden, dh cluster1.fa und cluster2.fa .
Weitere Informationen finden Sie unter -h/--help für weitere Informationen:
usage: clinker [-h] [--version] [-r RANGES [RANGES ...]] [-gf GENE_FUNCTIONS] [-na] [-i IDENTITY] [-j JOBS] [-s SESSION] [-ji JSON_INDENT] [-f] [-o OUTPUT] [-p [PLOT]] [-dl DELIMITER] [-dc DECIMALS] [-hl] [-ha] [-mo MATRIX_OUT] [-ufo] [files ...]
clinker: Automatic creation of publication-ready gene cluster comparison figures.
clinker generates gene cluster comparison figures from GenBank files. It performs pairwise local or global alignments between every sequence in every unique pair of clusters and generates interactive, to-scale comparison figures using the clustermap.js library.
optional arguments:
-h, --help show this help message and exit
--version show program's version number and exit
Input options:
files Gene cluster GenBank files
-r RANGES [RANGES ...], --ranges RANGES [RANGES ...]
Scaffold extraction ranges. If a range is specified, only features within the range will be extracted from the scaffold. Ranges should be formatted like: scaffold:start-end (e.g. scaffold_1:15000-40000)
-gf GENE_FUNCTIONS, --gene_functions GENE_FUNCTIONS
2-column CSV file containing gene functions, used to build gene groups from same function instead of sequence similarity (e.g. GENE_001,PKS-NRPS).
Alignment options:
-na, --no_align Do not align clusters
-i IDENTITY, --identity IDENTITY
Minimum alignment sequence identity [default: 0.3]
-j JOBS, --jobs JOBS Number of alignments to run in parallel (0 to use the number of CPUs) [default: 0]
Output options:
-s SESSION, --session SESSION
Path to clinker session
-ji JSON_INDENT, --json_indent JSON_INDENT
Number of spaces to indent JSON [default: none]
-f, --force Overwrite previous output file
-o OUTPUT, --output OUTPUT
Save alignments to file
-p [PLOT], --plot [PLOT]
Plot cluster alignments using clustermap.js. If a path is given, clinker will generate a portable HTML file at that path. Otherwise, the plot will be served dynamically using Python's HTTP server.
-dl DELIMITER, --delimiter DELIMITER
Character to delimit output by [default: human readable]
-dc DECIMALS, --decimals DECIMALS
Number of decimal places in output [default: 2]
-hl, --hide_link_headers
Hide alignment column headers
-ha, --hide_aln_headers
Hide alignment cluster name headers
-mo MATRIX_OUT, --matrix_out MATRIX_OUT
Save cluster similarity matrix to file
Visualisation options:
-ufo, --use_file_order
Display clusters in order of input files
Example usage
-------------
Align clusters, plot results and print scores to screen:
$ clinker files/*.gbk
Only save gene-gene links when identity is over 50%:
$ clinker files/*.gbk -i 0.5
Save an alignment session for later:
$ clinker files/*.gbk -s session.json
Save alignments to file, in comma-delimited format, with 4 decimal places:
$ clinker files/*.gbk -o alignments.csv -dl "," -dc 4
Generate visualisation:
$ clinker files/*.gbk -p
Save visualisation as a static HTML document:
$ clinker files/*.gbk -p plot.html
Cameron Gilchrist, 2020
Standardmäßig weist es Clinker automatisch einen Namen und eine Farbe für jede Gruppe homologen Gene zu. Sie können stattdessen Namen (dh Funktionen) mit dem Argument -gf/--gene_functions vorab, in dem eine 2-Spal-Comma-getrennte Datei vorliegt, wie:
GENE_001,Cytochrome P450
GENE_002,Cytochrome P450
GENE_003,Methyltransferase
GENE_004,Methyltransferase
Dies erzeugt zwei Gruppen, Cytochrom P450 (Gene_001 und 002) und Methyltransferase (Gene_003, Gene_004). Wenn andere homologe Gene identifiziert werden, werden diese Gruppen automatisch hinzugefügt.
Ab CLINKER V0.0.28 können Sie jetzt Farben für Gene angeben, die durch das Argument -gf/--gene_functions definiert sind. Verwenden Sie dazu das Argument -cm/--colour_map , das auch eine 2-Spalte-CSV-Datei mit dem Gruppennamen und dem hexadezimalen Farbcode enthält wie:
Cytochrome P450,#FF0000
Methyltransferase,#0000FF


