clinker
v0.0.30
Tanto Cblaster como Clinker ahora se pueden usar sin instalación en el servidor web Cagecat.
General de comparación de clúster Generador
Clinker es una tubería para generar fácilmente cifras de comparación de clúster de genes de calidad de publicación.
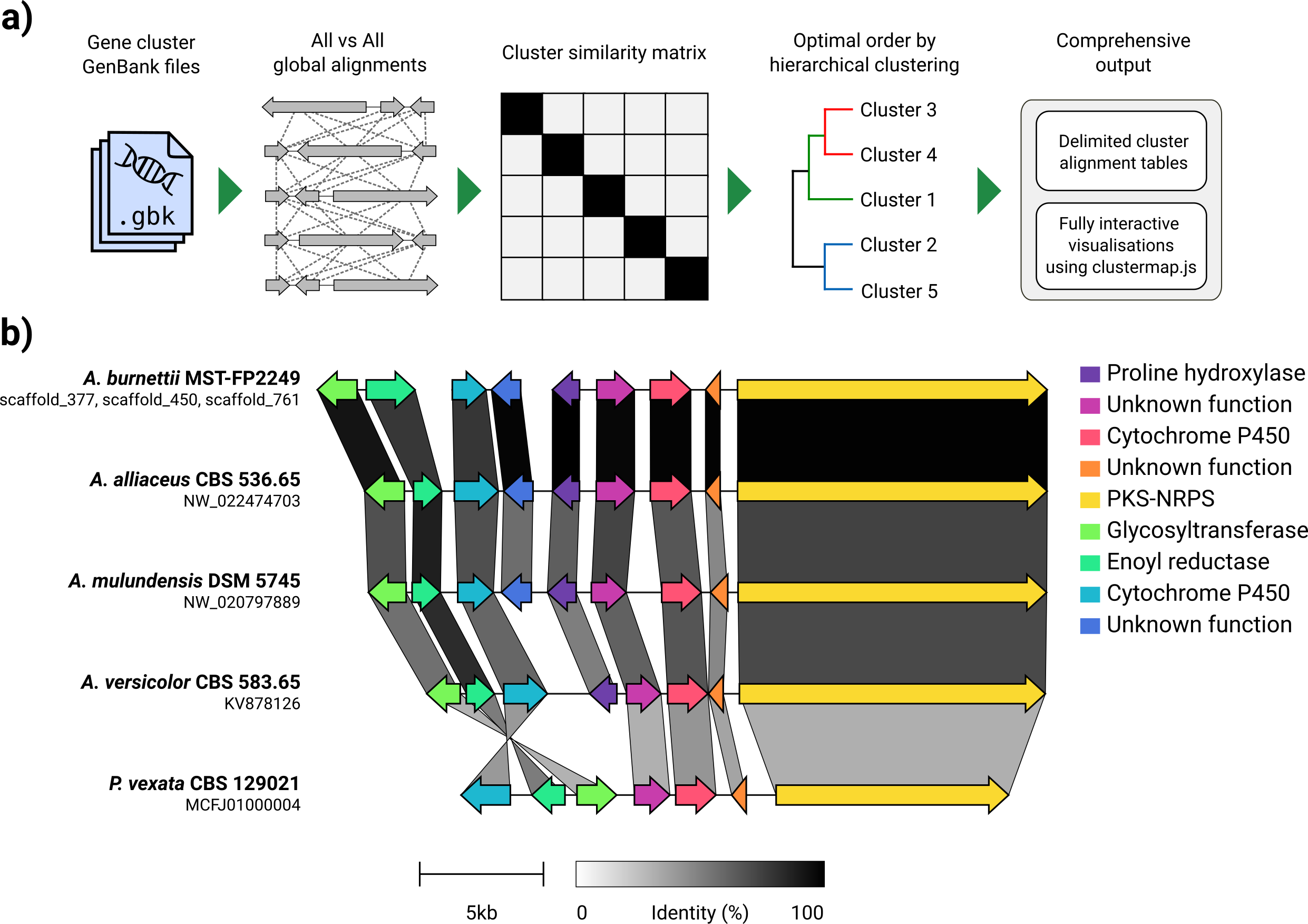
Dado un conjunto de archivos de genBank, Clinker extraerá automáticamente las traducciones de proteínas, realizará alineaciones globales entre secuencias en cada clúster, determinará el orden de visualización óptimo basado en la similitud de clúster y generará una visualización interactiva (usando clustermap.js) que se puede ajustar extensamente antes de ser exportado como un archivo SVG.
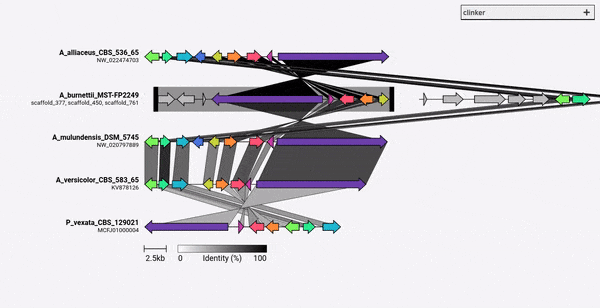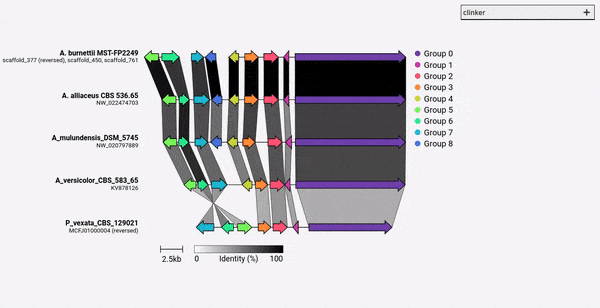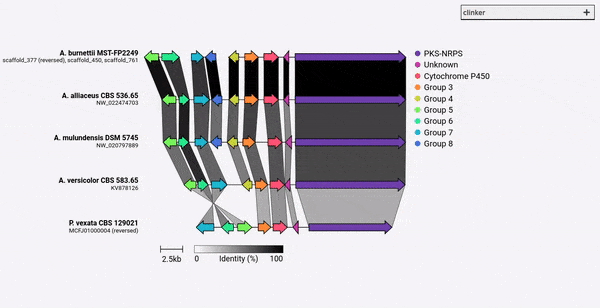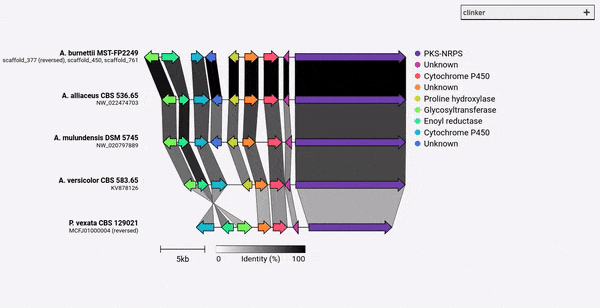
Clinker fue diseñado principalmente como una forma simple de visualizar grupos de grupos de genes biosintéticos homólogos, que típicamente son regiones genómicas pequeñas con no muchos genes (como en el ejemplo GIF). Realiza alineaciones por pares de todos los genes en todos los archivos de entrada utilizando el alineador integrado en Biopython, luego genera un documento SVG interactivo en el navegador. La etapa de alineación se escalará muy mal a múltiples genomas con muchos genes, y la visualización resultante también será muy lenta dada la cantidad de elementos SVG que contendrá. Si está buscando alinear genomas completos, es probable que se sirva mejor utilizando herramientas construidas para ese propósito (por ejemplo, cactus).
Clinker se puede instalar directamente a través de PIP:
pip install clinker
Clonando el código fuente de GitHub:
git clone https://github.com/gamcil/clinker.git
cd clinker
pip install .
O, a través de conda:
conda create -n clinker -c conda-forge -c bioconda clinker-py
conda activate clinker
Si encontró útil Clinker, cite:
clinker & clustermap.js: Automatic generation of gene cluster comparison figures.
Gilchrist, C.L.M., Chooi, Y.-H., 2020.
Bioinformatics. doi: https://doi.org/10.1093/bioinformatics/btab007
Correr clinker puede ser tan simple como:
clinker clusters/*.gbk
Esto se leerá en todos los archivos de GenBank dentro de la carpeta, los alineará e imprimirá las alineaciones en el terminal. Para generar la visualización, use el argumento -p/--plot :
clinker clusters/*.gbk -p <optional: file name to save static HTML>
Clinker también puede analizar archivos GFF3:
clinker cluster1.gff3 cluster2.gff3 -p
Nota: Un archivo FASTA correspondiente del mismo nombre (Extensiones ".fa", ".fsa", ".fna", ".Fasta" o ".faa") debe encontrarse en el mismo directorio que el GFF3, es decir, cluster1.fa y cluster2.fa .
Ver -h/--help para más información:
usage: clinker [-h] [--version] [-r RANGES [RANGES ...]] [-gf GENE_FUNCTIONS] [-na] [-i IDENTITY] [-j JOBS] [-s SESSION] [-ji JSON_INDENT] [-f] [-o OUTPUT] [-p [PLOT]] [-dl DELIMITER] [-dc DECIMALS] [-hl] [-ha] [-mo MATRIX_OUT] [-ufo] [files ...]
clinker: Automatic creation of publication-ready gene cluster comparison figures.
clinker generates gene cluster comparison figures from GenBank files. It performs pairwise local or global alignments between every sequence in every unique pair of clusters and generates interactive, to-scale comparison figures using the clustermap.js library.
optional arguments:
-h, --help show this help message and exit
--version show program's version number and exit
Input options:
files Gene cluster GenBank files
-r RANGES [RANGES ...], --ranges RANGES [RANGES ...]
Scaffold extraction ranges. If a range is specified, only features within the range will be extracted from the scaffold. Ranges should be formatted like: scaffold:start-end (e.g. scaffold_1:15000-40000)
-gf GENE_FUNCTIONS, --gene_functions GENE_FUNCTIONS
2-column CSV file containing gene functions, used to build gene groups from same function instead of sequence similarity (e.g. GENE_001,PKS-NRPS).
Alignment options:
-na, --no_align Do not align clusters
-i IDENTITY, --identity IDENTITY
Minimum alignment sequence identity [default: 0.3]
-j JOBS, --jobs JOBS Number of alignments to run in parallel (0 to use the number of CPUs) [default: 0]
Output options:
-s SESSION, --session SESSION
Path to clinker session
-ji JSON_INDENT, --json_indent JSON_INDENT
Number of spaces to indent JSON [default: none]
-f, --force Overwrite previous output file
-o OUTPUT, --output OUTPUT
Save alignments to file
-p [PLOT], --plot [PLOT]
Plot cluster alignments using clustermap.js. If a path is given, clinker will generate a portable HTML file at that path. Otherwise, the plot will be served dynamically using Python's HTTP server.
-dl DELIMITER, --delimiter DELIMITER
Character to delimit output by [default: human readable]
-dc DECIMALS, --decimals DECIMALS
Number of decimal places in output [default: 2]
-hl, --hide_link_headers
Hide alignment column headers
-ha, --hide_aln_headers
Hide alignment cluster name headers
-mo MATRIX_OUT, --matrix_out MATRIX_OUT
Save cluster similarity matrix to file
Visualisation options:
-ufo, --use_file_order
Display clusters in order of input files
Example usage
-------------
Align clusters, plot results and print scores to screen:
$ clinker files/*.gbk
Only save gene-gene links when identity is over 50%:
$ clinker files/*.gbk -i 0.5
Save an alignment session for later:
$ clinker files/*.gbk -s session.json
Save alignments to file, in comma-delimited format, with 4 decimal places:
$ clinker files/*.gbk -o alignments.csv -dl "," -dc 4
Generate visualisation:
$ clinker files/*.gbk -p
Save visualisation as a static HTML document:
$ clinker files/*.gbk -p plot.html
Cameron Gilchrist, 2020
Por defecto, Clinker asigna automáticamente un nombre y color para cada grupo de genes homólogos. En su lugar, puede usar los nombres previos al asignación (funciones de es decir) usando el argumento -gf/--gene_functions , que toma un archivo separado por comas de 2 columnas como:
GENE_001,Cytochrome P450
GENE_002,Cytochrome P450
GENE_003,Methyltransferase
GENE_004,Methyltransferase
Esto generará dos grupos, el citocromo P450 (Gene_001 y 002) y la metiltransferasa (Gene_003, Gene_004). Si se identifican otros genes homólogos, se agregarán automáticamente a estos grupos.
A partir de Clinker V0.0.28, ahora puede especificar colores para genes definidos por el argumento -gf/--gene_functions . Para hacer esto, use el argumento -cm/--colour_map que también toma un archivo CSV de 2 columnas que contiene el nombre del grupo y el código de color hexadecimal como:
Cytochrome P450,#FF0000
Methyltransferase,#0000FF


