DiffDock Pocket
Additional Data
DiffDock-Pocket est un modèle d'amarrage entièrement atome fonctionnant au niveau de la poche. Il met en œuvre la diffusion sur les angles de torsion de la chaîne latérale et est optimisé pour les structures générées par calcul. Dans ce référentiel, vous trouverez le code pour former un modèle, exécuter l'inférence, les visualisations et les poids que nous avons utilisés pour réaliser les nombres présentés dans l'article. Ce référentiel est à l'origine une fourche de DiffDock, donc certaines des commandes peuvent sembler familières - mais elle a été étendue, adaptée et modifiée dans tant d'endroits que vous ne pouvez pas vous attendre à une compatibilité des deux programmes. Soyez conscient et ne mélangez pas ces deux programmes!
Si vous voulez un moyen facile d'exécuter l'inférence, vous pouvez utiliser l'interface HuggingFace ici.
N'hésitez pas à créer des problèmes ou des PRS si vous avez des problèmes avec ce référentiel! Veuillez considérer le code de conduite.
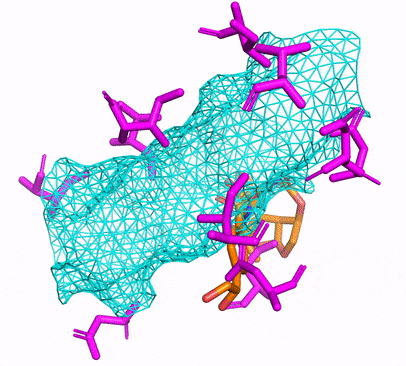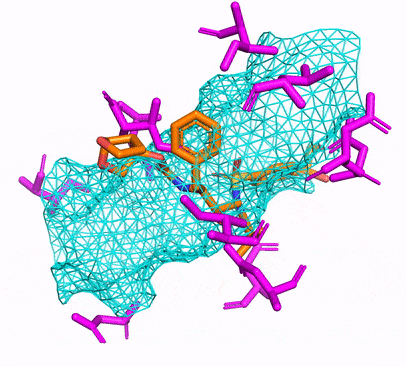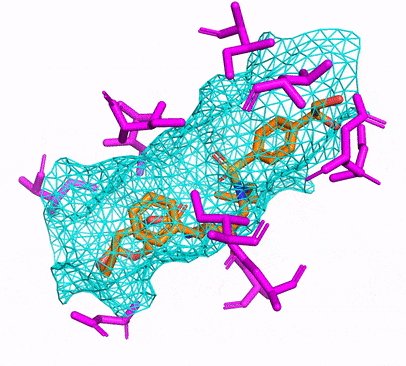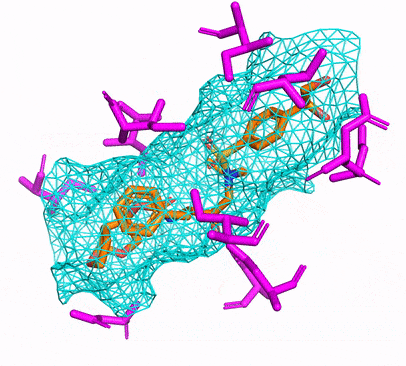
Vous pouvez créer l'environnement CPU ou CUDA Conda en exécutant simplement
conda env create -f environment[-cpu].yml
avec ou sans le drapeau CPU. La version CUDA est recommandée, car elle est beaucoup plus rapide.
Dans cet exemple minimal, nous utilisons le 3dpf complexe et remettons le ligand avec des cahins latéraux flexibles. Nous dessions 40 échantillons et les classons avec le modèle de confiance. Toutes les prédictions seront stockées dans results/ .
python inference.py --protein_path example_data/3dpf_protein.pdb --ligand example_data/3dpf_ligand.sdf --batch_size 20 --samples_per_complex 40 --keep_local_structures --save_visualisation
Ici, nous avons spécifié l'indicateur --keep_local_structures pour ne pas modifier le ligand. Avec cela, la poche et les chaînes latérales pour modéliser flexible sont automatiquement déterminées.
Si cette commande manque de mémoire, vous pouvez diminuer le paramètre --batch_size ou --samples_per_complex . Abaissement --batch_size n'a pas d'impact sur la qualité des résultats, mais augmentera le temps d'exécution.
Une commande similaire pour les structures générées par calcul serait:
python inference.py --protein_path example_data/3dpf_protein_esm.pdb --ligand example_data/3dpf_ligand.sdf --batch_size 20 --samples_per_complex 40 --keep_local_structures --save_visualisation
Pour effectuer une minimisation d'énergie sur la pose de ligand prédite supérieure, vous pouvez utiliser l'indicateur --relax .
Si vous connaissez votre poche de liaison et les chaînes latérales à modéliser comme flexibles, vous pouvez les spécifier. Dans ce cas, vous pouvez également laisser tomber --keep_local_structures . Nous conseillons d'utiliser notre modèle fourni uniquement avec des acides aminés flexibles qui ont au moins un atome lourd à moins de 3,5 A de tout atome de ligand lourd. Cela empêche les données hors distribution.
python inference.py --protein_path example_data/3dpf_protein.pdb --ligand example_data/3dpf_ligand.sdf --batch_size 20 --samples_per_complex 40 --save_visualisation --pocket_center_x 9.7742 --pocket_center_y 27.2863 --pocket_center_z 14.6573 --flexible_sidechains A:160-A:193-A:197-A:198-A:222-A:224-A:227
Ou si vous n'avez pas de ligand à portée de main, vous pouvez également utiliser la représentation des sourires.
--ligand [H]/N=C1/C(=O)C(=O)[C@@]1([H])[N@@+]1([H])C([H])([H])c2c([H])c(C([H])([H])N([H])C(=O)c3nc4sc5c(c4c(=O)n3[H])C([H])([H])C([H])([H])S(=O)(=O)C5([H])[H])c([H])c([H])c2C([H])([H])C1([H])[H]
Cependant, dans ce cas, vous devez spécifier votre centre de poche et vos chaînes latérales flexibles.
Si vous traitez avec plusieurs complexes, jetez un œil à data/protein_ligand_example_csv avec le Flag --protein_ligand_csv data/protein_ligand_example_csv.csv . Cela vous permet de spécifier l'inférence sur plusieurs complexes à la fois, où vous pouvez spécifier le centre de poche et les chaînes latérales flexibles pour chaque complexe.
Lors du choix du centre de poche, il est préférable de déterminer les acides aminés qui sont proches du site de liaison et de calculer la moyenne de leurs coordonnées C-alpha. Dans notre formation, nous avons choisi tous les acides aminés où leur atome c-alpha se trouve à moins de 5 ans de tout atome lourd du ligand.
Nous utilisons PDBBIND que vous pouvez télécharger à partir de nombreuses sources. Cependant, nous fournissons également un ensemble de données PDBBIND pré-traité ici que vous pouvez déballer avec unzip . Toutes les protéines ont été fixées avec PDBFixer et alignées pour minimiser le RMSD dans la poche. Décompressez-le sur data/PDBBIND_atomCorrected .
Former le modèle grand score:
python -m train --run_name big_score_model --test_sigma_intervals --log_dir workdir --lr 1e-3 --tr_sigma_min 0.1 --tr_sigma_max 5 --rot_sigma_min 0.03 --rot_sigma_max 1.55 --tor_sigma_min 0.03 --sidechain_tor_sigma_min 0.03 --batch_size 16 --ns 60 --nv 10 --num_conv_layers 6 --distance_embed_dim 64 --cross_distance_embed_dim 64 --sigma_embed_dim 64 --dynamic_max_cross --scheduler plateau --scale_by_sigma --dropout 0.1 --sampling_alpha 1 --sampling_beta 1 --remove_hs --c_alpha_max_neighbors 24 --atom_max_neighbors 8 --receptor_radius 15 --num_dataloader_workers 1 --cudnn_benchmark --rot_alpha 1 --rot_beta 1 --tor_alpha 1 --tor_beta 1 --val_inference_freq 5 --use_ema --scheduler_patience 30 --n_epochs 750 --all_atom --sh_lmax 1 --split_train data/splits/timesplit_no_lig_overlap_train --split_val data/splits/timesplit_no_lig_overlap_val_aligned --pocket_reduction --pocket_buffer 10 --flexible_sidechains --flexdist 3.5 --flexdist_distance_metric prism --protein_file protein_esmfold_aligned_tr_fix --compare_true_protein --conformer_match_sidechains --conformer_match_score exp --match_max_rmsd 2 --use_original_conformer_fallback --use_original_conformer
Les poids du modèle sont enregistrés dans le répertoire workdir et peuvent être utilisés pour l'inférence.
Pour former le modèle de confiance, vous pouvez utiliser le modèle de score précédemment formé pour générer les échantillons. Cependant, nous avons choisi de former un autre modèle de score (plus petit) avec une traduction maximale plus élevée, de sorte que le modèle de confiance voit des échantillons plus diversifiés et que les données d'entraînement peuvent être produites plus rapidement.
Pour entraîner ce modèle de score plus petit avec une traduction maximale plus élevée, exécutez ce qui suit:
python -m train --run_name small_score_model --test_sigma_intervals --esm_embeddings_path data/esm2_3billion_embeddings.pt --cache_path data/cache --log_dir workdir --lr 1e-3 --tr_sigma_min 0.1 --tr_sigma_max 15 --rot_sigma_min 0.03 --rot_sigma_max 1.55 --tor_sigma_min 0.03 --sidechain_tor_sigma_min 0.03 --batch_size 16 --ns 32 --nv 6 --num_conv_layers 5 --dynamic_max_cross --scheduler plateau --scale_by_sigma --dropout 0.1 --sampling_alpha 1 --sampling_beta 1 --remove_hs --c_alpha_max_neighbors 24 --atom_max_neighbors 12 --receptor_radius 15 --num_dataloader_workers 1 --cudnn_benchmark --rot_alpha 1 --rot_beta 1 --tor_alpha 1 --tor_beta 1 --val_inference_freq 5 --use_ema --scheduler_patience 30 --n_epochs 500 --all_atom --sh_lmax 1 --split_train data/splits/timesplit_no_lig_overlap_train --split_val data/splits/timesplit_no_lig_overlap_val_aligned --pocket_reduction --pocket_buffer 10 --flexible_sidechains --flexdist 3.5 --flexdist_distance_metric prism --protein_file protein_esmfold_aligned_tr_fix --compare_true_protein --conformer_match_sidechains --conformer_match_score exp --match_max_rmsd 2 --use_original_conformer_fallback --use_original_conformer
Le modèle de score utilisé pour générer les échantillons pour former le modèle de confiance ne doit pas être le même que le modèle de score utilisé avec ce modèle de confiance pendant l'inférence.
Former le modèle de confiance en exécutant ce qui suit:
python -m filtering.filtering_train --run_name confidence_model --original_model_dir workdir/small_score_model --ckpt best_ema_inference_epoch_model.pt --inference_steps 20 --samples_per_complex 7 --batch_size 16 --n_epochs 100 --lr 3e-4 --scheduler_patience 50 --ns 24 --nv 6 --num_conv_layers 5 --dynamic_max_cross --scale_by_sigma --dropout 0.1 --all_atoms --sh_lmax 1 --split_train data/splits/timesplit_no_lig_overlap_train --split_val data/splits/timesplit_no_lig_overlap_val_aligned --log_dir workdir --cache_path .cache/data_filtering data/PDBBIND_atomCorrected --remove_hs --c_alpha_max_neighbors 24 --receptor_radius 15 --esm_embeddings_path data/esm2_3billion_embeddings.pt --main_metric loss --main_metric_goal min --best_model_save_frequency 5 --rmsd_classification_cutoff 2 --sc_rmsd_classification_cutoff 1 --protein_file protein_esmfold_aligned_tr_fix --use_original_model_cache --pocket_reduction --pocket_buffer 10 --cache_creation_id 1 --cache_ids_to_combine 1 2 3 4
d'abord avec --cache_creation_id 1 alors --cache_creation_id 2 etc. jusqu'à 4
Maintenant, tout est formé et vous pouvez faire en sorte que votre nouveau modèle :).
Si vous utilisez DiffDock-Pocket dans votre recherche, veuillez citer l'article suivant:
@article{plainer2023diffdockpocket,
author = {Plainer, Michael and Toth, Marcella and Dobers, Simon and St{"a}rk, Hannes and Corso, Gabriele and Marquet, C{'e}line and Barzilay, Regina},
title = {{DiffDock-Pocket}: Diffusion for Pocket-Level Docking with Sidechain Flexibility},
year = {2023},
maintitle = {Advances in Neural Information Processing Systems},
booktitle = {Machine Learning in Structural Biology},
}
Merci @jsilter d'avoir rendu ce code plus facile à utiliser et de créer l'interface HuggingFace!


