DiffDock Pocket
Additional Data
Diffdock-Pocket ist ein All-Atom-Docking-Modell, das auf Taschenebene arbeitet. Es implementiert die Diffusion über Seitenketten -Torsionswinkel und wird für rechnerisch erzeugte Strukturen optimiert. In diesem Repository finden Sie den Code, um ein Modell zu trainieren, Inferenz, Visualisierungen und die Gewichte auszuführen, mit denen wir die im Papier dargestellten Zahlen erreicht haben. Dieses Repository ist ursprünglich eine Gabel aus Diffdock, sodass einige der Befehle vertraut zu sein scheinen - aber an so vielen Stellen wurde es erweitert, angepasst und verändert, dass Sie keine Kompatibilität der beiden Programme erwarten können. Seien Sie sich bewusst und mischen Sie diese beiden Programme nicht!
Wenn Sie eine einfache Möglichkeit haben möchten, Inferenz auszuführen, können Sie hier die Schnittstelle für das Suggingface verwenden.
Fühlen Sie sich frei, Probleme oder PRs zu erstellen, wenn Sie Probleme mit diesem Repository haben! Bitte betrachten Sie den Verhaltenskodex.
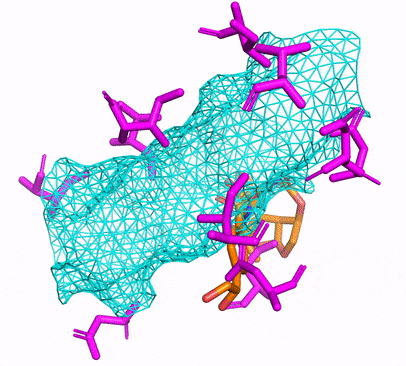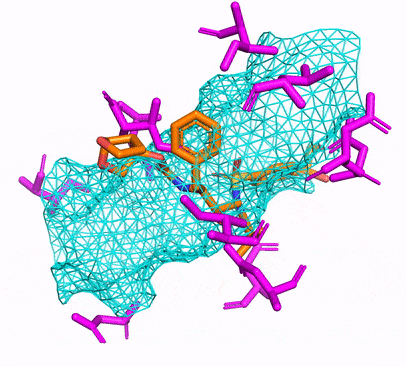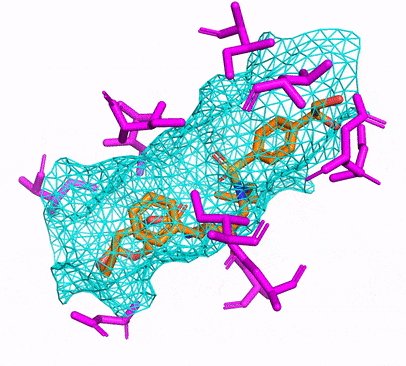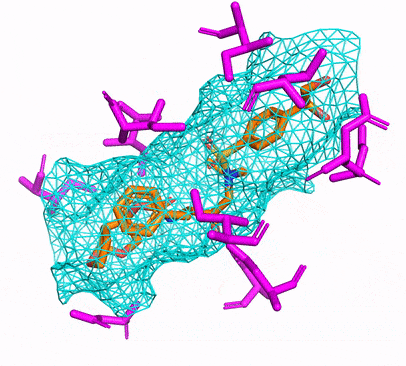
Sie können entweder die CPU- oder CUDA Conda -Umgebung erstellen, indem Sie einfach laufen
conda env create -f environment[-cpu].yml
mit oder ohne die CPU -Flagge. Die CUDA -Version wird empfohlen, da sie viel schneller ist.
In diesem minimalen Beispiel verwenden wir den komplexen 3dpf und dockten den Liganden mit flexiblen Seitencahinen neu. Wir ziehen 40 Proben und bewerten sie mit dem Konfidenzmodell. Alle Vorhersagen werden in results/ .
python inference.py --protein_path example_data/3dpf_protein.pdb --ligand example_data/3dpf_ligand.sdf --batch_size 20 --samples_per_complex 40 --keep_local_structures --save_visualisation
Hier haben wir das Flag --keep_local_structures angegeben, um den Liganden nicht zu ändern. Damit werden die Tasche und welche Seitenketten flexibel modellieren.
Wenn dieser Befehl aus dem Speicher ausgeht, können Sie den Parameter --batch_size oder den Parameter --samples_per_complex verringern. Absenkung --batch_size hat keinen Einfluss auf die Qualität der Ergebnisse, sondern erhöht die Laufzeit.
Ein ähnlicher Befehl für rechnerisch erzeugte Strukturen wäre:
python inference.py --protein_path example_data/3dpf_protein_esm.pdb --ligand example_data/3dpf_ligand.sdf --batch_size 20 --samples_per_complex 40 --keep_local_structures --save_visualisation
Um die Energieminimierung in der obersten 1-vorhergesagten Ligandenpose durchzuführen, können Sie das Flag --relax Flag verwenden.
Wenn Sie Ihre Bindungstasche kennen und welche Seitenketten als flexibel modellieren sollen, können Sie diese angeben. In diesem Fall können Sie auch fallen --keep_local_structures . Wir empfehlen, unser bereitgestelltes Modell nur mit flexiblen Aminosäuren zu verwenden, die mindestens ein schweres Atom innerhalb von 3,5a eines schweren Ligandenatoms haben. Dies verhindert Daten außerhalb der Verteilung.
python inference.py --protein_path example_data/3dpf_protein.pdb --ligand example_data/3dpf_ligand.sdf --batch_size 20 --samples_per_complex 40 --save_visualisation --pocket_center_x 9.7742 --pocket_center_y 27.2863 --pocket_center_z 14.6573 --flexible_sidechains A:160-A:193-A:197-A:198-A:222-A:224-A:227
Oder wenn Sie keinen Liganden zur Hand haben, können Sie auch die Smiles -Darstellung verwenden.
--ligand [H]/N=C1/C(=O)C(=O)[C@@]1([H])[N@@+]1([H])C([H])([H])c2c([H])c(C([H])([H])N([H])C(=O)c3nc4sc5c(c4c(=O)n3[H])C([H])([H])C([H])([H])S(=O)(=O)C5([H])[H])c([H])c([H])c2C([H])([H])C1([H])[H]
In diesem Fall müssen Sie jedoch Ihre Taschenmitte und flexible Seitenketten angeben.
Wenn Sie sich mit mehreren Komplexen befassen, schauen Sie sich data/protein_ligand_example_csv mit dem Flag --protein_ligand_csv data/protein_ligand_example_csv.csv an. Auf diese Weise können Sie Inferenz in mehreren Komplexen gleichzeitig angeben, in denen Sie für jeden Komplex die Taschenmitte und flexible Seitenketten angeben können.
Bei der Auswahl des Taschenzentrums ist es am besten, die Aminosäuren zu bestimmen, die sich in der Nähe der Bindungsstelle befinden und den Mittelwert ihrer C-Alpha-Koordinaten berechnen. In unserem Training haben wir alle Aminosäuren ausgewählt, in denen sich ihr C-Alpha-Atom innerhalb von 5a eines schweren Atoms des Liganden befindet.
Wir verwenden PDBBind, die Sie aus vielen Quellen herunterladen können. Wir bieten jedoch auch einen vorverarbeiteten PDBBind-Datensatz hier, den Sie mit unzip auspacken können. Alle Proteine wurden mit PDBFixer fixiert und ausgerichtet, um die RMSD in der Tasche zu minimieren. Packen Sie es auf data/PDBBIND_atomCorrected aus.
Trainieren Sie das große Score -Modell:
python -m train --run_name big_score_model --test_sigma_intervals --log_dir workdir --lr 1e-3 --tr_sigma_min 0.1 --tr_sigma_max 5 --rot_sigma_min 0.03 --rot_sigma_max 1.55 --tor_sigma_min 0.03 --sidechain_tor_sigma_min 0.03 --batch_size 16 --ns 60 --nv 10 --num_conv_layers 6 --distance_embed_dim 64 --cross_distance_embed_dim 64 --sigma_embed_dim 64 --dynamic_max_cross --scheduler plateau --scale_by_sigma --dropout 0.1 --sampling_alpha 1 --sampling_beta 1 --remove_hs --c_alpha_max_neighbors 24 --atom_max_neighbors 8 --receptor_radius 15 --num_dataloader_workers 1 --cudnn_benchmark --rot_alpha 1 --rot_beta 1 --tor_alpha 1 --tor_beta 1 --val_inference_freq 5 --use_ema --scheduler_patience 30 --n_epochs 750 --all_atom --sh_lmax 1 --split_train data/splits/timesplit_no_lig_overlap_train --split_val data/splits/timesplit_no_lig_overlap_val_aligned --pocket_reduction --pocket_buffer 10 --flexible_sidechains --flexdist 3.5 --flexdist_distance_metric prism --protein_file protein_esmfold_aligned_tr_fix --compare_true_protein --conformer_match_sidechains --conformer_match_score exp --match_max_rmsd 2 --use_original_conformer_fallback --use_original_conformer
Die Modellgewichte werden im workdir -Verzeichnis gespeichert und können für die Inferenz verwendet werden.
Um das Konfidenzmodell zu trainieren, können Sie das zuvor trainierte Score -Modell verwenden, um die Proben zu generieren. Wir haben uns jedoch entschieden, ein anderes (kleineres) Score -Modell mit einer höheren maximalen Ligandenübersetzung zu trainieren, so dass das Konfidenzmodell vielfältigere Proben sieht und die Trainingsdaten schneller erzeugt werden können.
Führen Sie Folgendes aus:
python -m train --run_name small_score_model --test_sigma_intervals --esm_embeddings_path data/esm2_3billion_embeddings.pt --cache_path data/cache --log_dir workdir --lr 1e-3 --tr_sigma_min 0.1 --tr_sigma_max 15 --rot_sigma_min 0.03 --rot_sigma_max 1.55 --tor_sigma_min 0.03 --sidechain_tor_sigma_min 0.03 --batch_size 16 --ns 32 --nv 6 --num_conv_layers 5 --dynamic_max_cross --scheduler plateau --scale_by_sigma --dropout 0.1 --sampling_alpha 1 --sampling_beta 1 --remove_hs --c_alpha_max_neighbors 24 --atom_max_neighbors 12 --receptor_radius 15 --num_dataloader_workers 1 --cudnn_benchmark --rot_alpha 1 --rot_beta 1 --tor_alpha 1 --tor_beta 1 --val_inference_freq 5 --use_ema --scheduler_patience 30 --n_epochs 500 --all_atom --sh_lmax 1 --split_train data/splits/timesplit_no_lig_overlap_train --split_val data/splits/timesplit_no_lig_overlap_val_aligned --pocket_reduction --pocket_buffer 10 --flexible_sidechains --flexdist 3.5 --flexdist_distance_metric prism --protein_file protein_esmfold_aligned_tr_fix --compare_true_protein --conformer_match_sidechains --conformer_match_score exp --match_max_rmsd 2 --use_original_conformer_fallback --use_original_conformer
Das Score -Modell, mit dem die Proben zum Training des Konfidenzmodells generiert werden, muss nicht das gleiche wie das Score -Modell sein, das während der Inferenz mit diesem Konfidenzmodell verwendet wird.
Trainieren Sie das Konfidenzmodell, indem Sie Folgendes ausführen:
python -m filtering.filtering_train --run_name confidence_model --original_model_dir workdir/small_score_model --ckpt best_ema_inference_epoch_model.pt --inference_steps 20 --samples_per_complex 7 --batch_size 16 --n_epochs 100 --lr 3e-4 --scheduler_patience 50 --ns 24 --nv 6 --num_conv_layers 5 --dynamic_max_cross --scale_by_sigma --dropout 0.1 --all_atoms --sh_lmax 1 --split_train data/splits/timesplit_no_lig_overlap_train --split_val data/splits/timesplit_no_lig_overlap_val_aligned --log_dir workdir --cache_path .cache/data_filtering data/PDBBIND_atomCorrected --remove_hs --c_alpha_max_neighbors 24 --receptor_radius 15 --esm_embeddings_path data/esm2_3billion_embeddings.pt --main_metric loss --main_metric_goal min --best_model_save_frequency 5 --rmsd_classification_cutoff 2 --sc_rmsd_classification_cutoff 1 --protein_file protein_esmfold_aligned_tr_fix --use_original_model_cache --pocket_reduction --pocket_buffer 10 --cache_creation_id 1 --cache_ids_to_combine 1 2 3 4
Zuerst mit --cache_creation_id 1 dann --cache_creation_id 2 usw. bis zu 4
Jetzt ist alles ausgebildet und Sie können mit Ihrem neuen Modell Schlussfolgerung führen :).
Wenn Sie Diffdock-Pocket in Ihrer Forschung verwenden, geben Sie bitte das folgende Papier an:
@article{plainer2023diffdockpocket,
author = {Plainer, Michael and Toth, Marcella and Dobers, Simon and St{"a}rk, Hannes and Corso, Gabriele and Marquet, C{'e}line and Barzilay, Regina},
title = {{DiffDock-Pocket}: Diffusion for Pocket-Level Docking with Sidechain Flexibility},
year = {2023},
maintitle = {Advances in Neural Information Processing Systems},
booktitle = {Machine Learning in Structural Biology},
}
Vielen Dank an @jsilter, dass Sie diesen Code leichter zu verwenden und die Schnittstelle für die Huggingface -Oberfläche zu erstellen!


