SCDC
1.0.0
SCDC เป็นวิธีการ deconvolution สำหรับ RNA-seq จำนวนมากที่ใช้ประโยชน์จากการแสดงออกของยีนเฉพาะประเภทเซลล์จาก ชุดข้อมูลอ้างอิง SCRA-seq หลายชุด SCDC ใช้วิธีการทั้งหมดในการรวมผลลัพธ์ deconvolution จากชุดข้อมูล SCRA-seq ที่แตกต่างกันซึ่งผลิตในห้องปฏิบัติการที่แตกต่างกันและในเวลาที่ต่างกัน
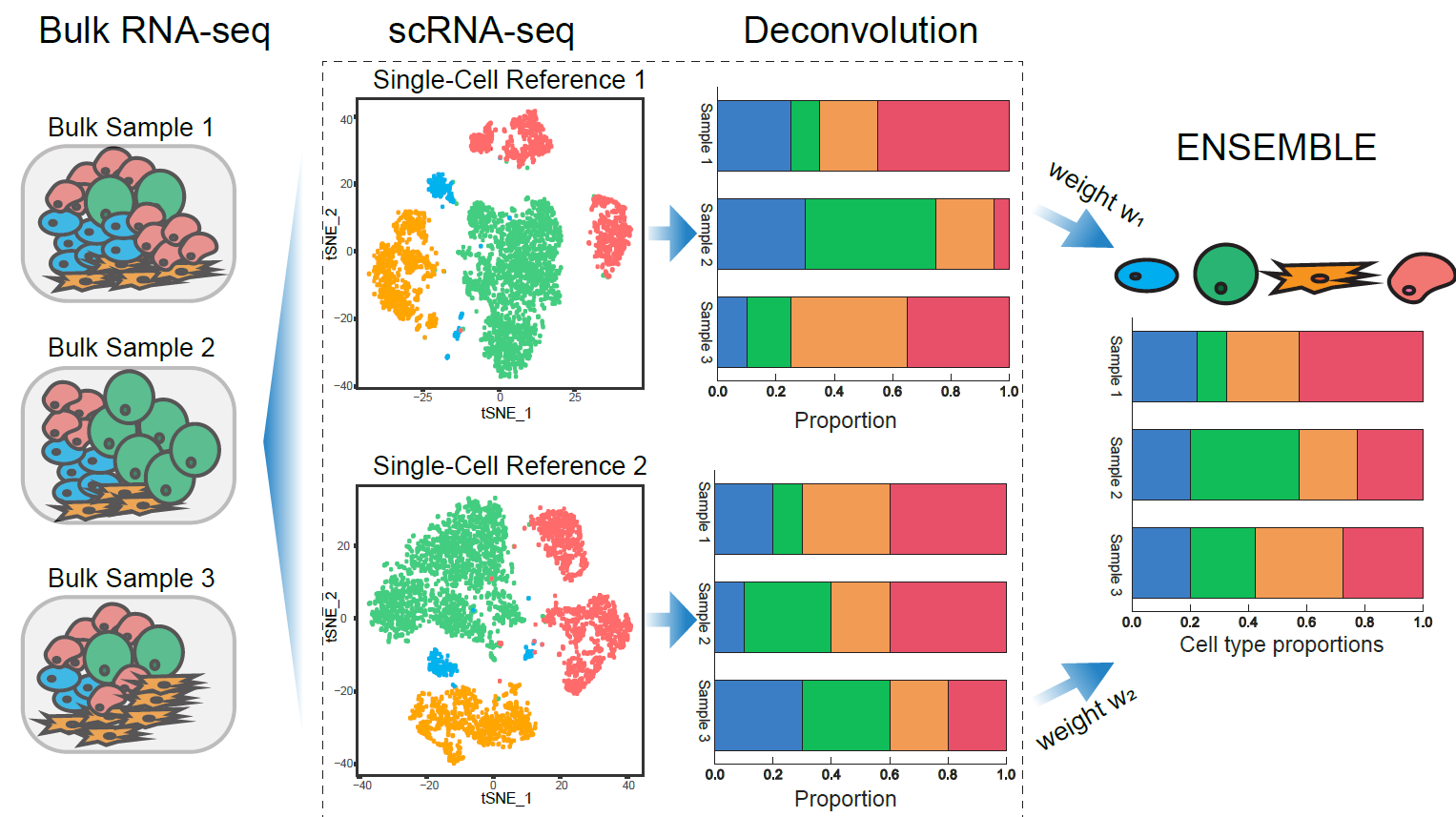
Meichen Dong, Aatish Thennavan, Eugene Urrutia, Yun Li, Charles M Perou, Fei Zou, Yuchao Jiang, SCDC: การแสดงออกของยีนจำนวนมาก deconvolution โดยการอ้างอิงลำดับ RNA ของเซลล์เดี่ยว org/10.1093/bib/bbz166
ใบอนุญาต: MIT
คุณสามารถติดตั้ง SCDC เวอร์ชันที่เผยแพร่จาก GitHub ด้วย:
ถ้า (! ต้องการ ("devtools")) {
Install.packages ("Devtools")
} devtools :: install_github ("meichendong/scdc"))ปัญหาแพ็คเกจการพึ่งพาเกี่ยวกับ 'xbioc' สามารถแก้ไขได้โดย:
Install.packages ("Remotes") Remotes :: Install_github ("Renozao/Xbioc")โปรดดูหน้า Vignettes
กระดาษ SCDC เผยแพร่ในการบรรยายสรุปในชีวสารสนเทศศาสตร์
คำถามเกี่ยวกับแพ็คเกจสามารถส่งอีเมลไปที่: [email protected]
เมื่อมีเพียง 'หัวข้อ/บุคคล' ในชุดข้อมูลเซลล์เดียวโปรดใช้ฟัง SCDC_qc_ONE() , SCDC_prop_ONE() ฟังก์ชั่น
แง่มุมที่อาจส่งผลกระทบต่อผลลัพธ์การสลายตัว:
รูปแบบข้อมูล: ตัวอย่างจำนวนมากและเซลล์เดี่ยวทั้งรูปแบบดิบ / รูปแบบเดียวกันหรือไม่? เราคาดว่ารูปแบบข้อมูลจะสอดคล้องและเทียบเคียงได้
การกรองยีน: คุณกรองยีน / ยีน ribosomal ที่แสดงออกต่ำหรือยีนไมโตคอนเดรียลหรือไม่? ยีนเหล่านี้อาจส่งผลกระทบต่อการวิเคราะห์ดาวน์สตรีม
ขนาดเซลล์และปัจจัยขนาดห้องสมุด: สำหรับเซลล์เดียวคุณคิดว่าผลรวมของการนับยีนทั้งหมด (ขนาดห้องสมุด) สามารถสะท้อนขนาดเซลล์ที่แท้จริงได้หรือไม่? นี่เป็นหนึ่งในสมมติฐานของเรา: อัตราส่วนของขนาดห้องสมุดระหว่างชนิดของเซลล์สามารถสะท้อนอัตราส่วนของขนาดเซลล์จริงระหว่างชนิดของเซลล์ ถ้าไม่คุณสามารถป้อนปัจจัยขนาดเซลล์ด้วยตนเองเมื่อสร้าง "พื้นฐานเมทริกซ์"
เซลล์ชนิดที่คล้ายกัน: มีเซลล์ประเภทที่อาจทำให้การวิเคราะห์สับสนหรือไม่? ตัวอย่างเช่นประเภทเซลล์ที่มีโปรไฟล์ /ยีนเครื่องหมายที่คล้ายกันมาก
ขาดประเภทเซลล์ที่สำคัญ / ปัญหาทางเทคนิค: คุณคาดหวังว่าขั้นตอนการเรียงลำดับจะสร้างความแตกต่างอย่างมากในกลุ่มและ SC แม้กระทั่งเทคนิคก็เหมือนกันหรือไม่? บางครั้งข้อมูลอ้างอิงเซลล์เดียวอาจสูญเสียข้อมูลสำหรับเซลล์บางประเภท ตัวอย่างเช่นมีเซลล์ไขมันในตัวอย่างจำนวนมากของคุณ แต่อย่างใดคุณก็ไม่มีข้อมูลเซลล์เดียว
Deconvolution โดยใช้ชุดข้อมูลอ้างอิงเดียว: คุณพยายามใช้ชุดข้อมูลอ้างอิงหนึ่งชุดเพื่อทดสอบว่าผลลัพธ์นั้นสมเหตุสมผลหรือไม่? ฉันเห็นคุณลอง Bisque คุณเคยลองวิธีอื่น ๆ เช่น Cibersortx หรือไม่? หากผลลัพธ์จากวิธีการ deconvolution "การอ้างอิงแบบเดียว" อื่น ๆ นั้นสมเหตุสมผลกว่าคุณสามารถป้อนข้อมูลเหล่านี้โดยตรงโดยใช้ขั้นตอนวงดนตรีของเรา


