SCDC
1.0.0
SCDC est une méthode de déconvolution pour l'ARN-seq en vrac qui exploite les expressions de gènes spécifiques de type cellulaire à partir de plusieurs ensembles de données de référence SCRNA-SEQ . SCDC adopte une méthode d'ensemble pour intégrer les résultats de déconvolution de différents ensembles de données SCRNA-SEQ qui sont produits dans différents laboratoires et à différents moments, traitant implicitement la confusion à effet par lots.
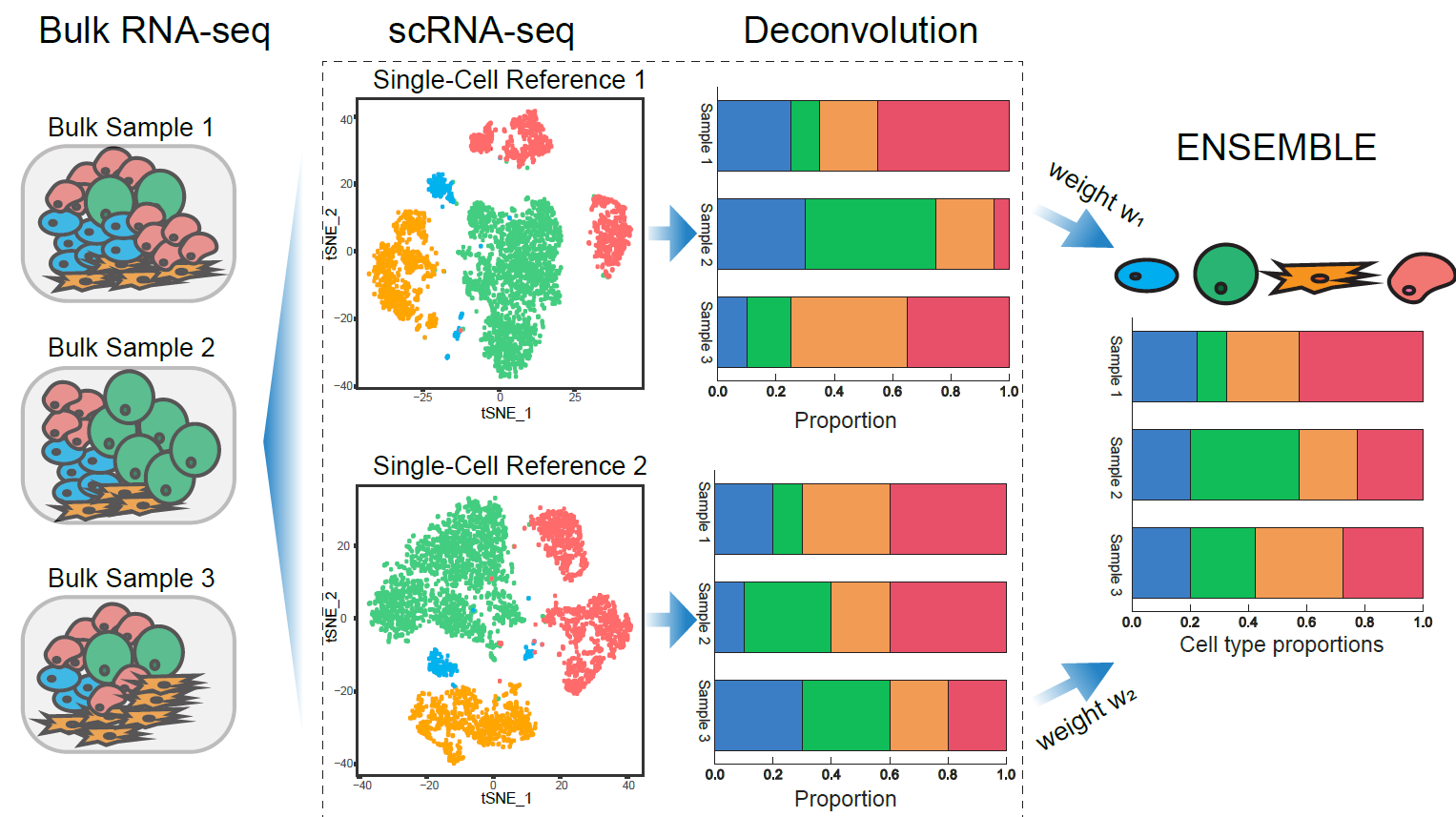
Meichen Dong, Aatish Thennavan, Eugene Urrutia, Yun Li, Charles M Perou, Fei Zou, Yuchao Jiang, SCDC: Bulk Gene Expression Deconvolution par plusieurs références de séquençage d'ARN à cellule unique, briefings en bioinformatique ,, BBZ166, https: // di. org / 10.1093 / bib / bbz166
Licence: MIT
Vous pouvez installer la version publiée de SCDC à partir de GitHub avec:
if (! require ("devtools")) {
install.packages ("Devtools")
} devtools :: install_github ("meichendong / scdc")Problème de package de dépendance concernant «xbioc» pourrait être résolu par:
install.packages ("Remotes") Remotes :: install_github ("renozao / xbioc")Veuillez consulter la page Vignettes.
Le document SCDC est publié lors d'information sur la bioinformatique.
Les questions concernant le package peuvent être envoyées par e-mail à: [email protected]
Lorsqu'il n'y a que «un sujet / individuel» dans l'ensemble de données à cellule unique, veuillez utiliser SCDC_qc_ONE() , SCDC_prop_ONE() .
Aspects qui pourraient affecter les résultats de la déconvolution:
Format de données: les échantillons en vrac et unique sont-ils à la fois des comptes bruts / même format? Nous nous attendons à ce que le format de données soit cohérent et comparable.
Filtrage des gènes: Avez-vous filmé des gènes / gènes ribosomaux / gènes mitochondriaux? Ces gènes peuvent affecter l'analyse en aval.
Taille des cellules et facteurs de taille de la bibliothèque: pour une seule cellule, pensez-vous que la somme de tous les dénombrements de gènes (la taille de la bibliothèque) pourrait refléter sa taille de cellule réelle? C'est l'une de nos hypothèses: le rapport des tailles de bibliothèque entre les types de cellules peut refléter le rapport des tailles de cellules réelles entre les types de cellules. Sinon, vous pouvez saisir manuellement le facteur de taille des cellules lors de la construction de la "matrice de base".
Des types de cellules similaires: existe-t-il des types de cellules qui pourraient potentiellement confondre l'analyse? Par exemple, les types de cellules qui ont des profils / gènes marqueurs très similaires.
Manquer les principaux types de cellules / problèmes techniques: vous attendez-vous à ce que la procédure de séquençage fasse une grande différence en vrac et SC même la technique est la même? Parfois, les données de référence à cellule unique peuvent perdre des informations pour certains types de cellules. Par exemple, il y a des cellules graisseuses dans vos échantillons en vrac, mais vous ne l'avez pas d'une manière ou d'une autre pour les données unicellulaires.
déconvolution à l'aide d'un ensemble de données de référence unique: avez-vous essayé d'utiliser un ensemble de données de référence pour tester si les résultats ont du sens généralement? Je vois que vous avez essayé la bisque. Avez-vous essayé d'autres méthodes comme Cibersortx? Si les résultats des autres méthodes de déconvolution «à une référence» ont plus de sens, vous pouvez les saisir directement en utilisant notre étape d'ensemble.


