DNABERT_2
1.0.0
repo에는 다음이 포함됩니다.
우리는 임베딩 공간에서 자연스럽게 다른 종의 게놈을 분리하고 분리하는 DNA 임베딩을 생성하기 위해 특별히 설계된 DNABERT-2를 기반으로 한 기초 모델 인 DNABERT-S를 게시합니다. 관심이 있으시면 여기에서 확인하십시오.
DNABERT-2
미리 훈련 된 모델은 huggingface에서 zhihan1996/DNABERT-2-117M 로 사용할 수 있습니다. Huggingface ModelHub에 대한 링크. 직접 다운로드 링크.
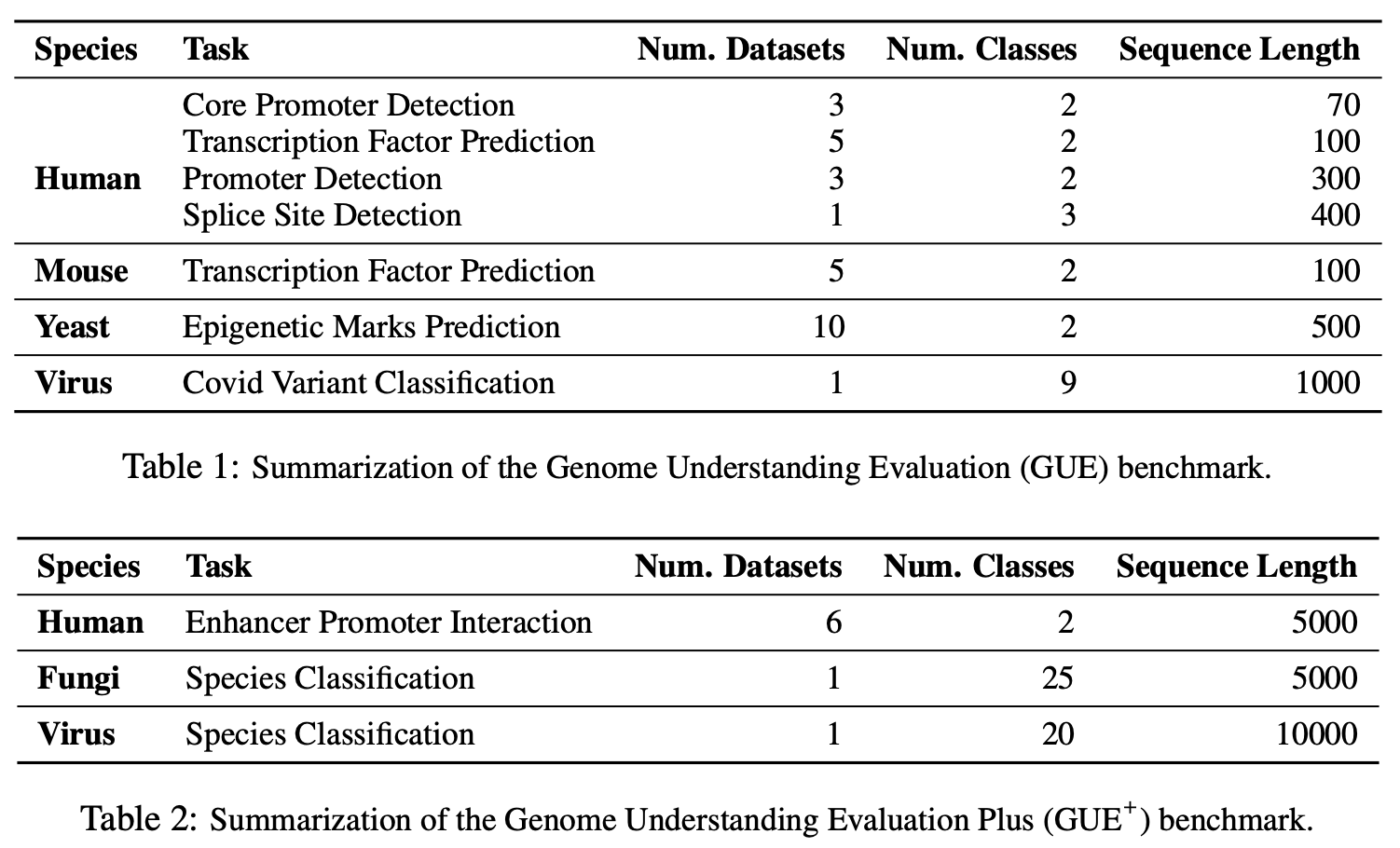
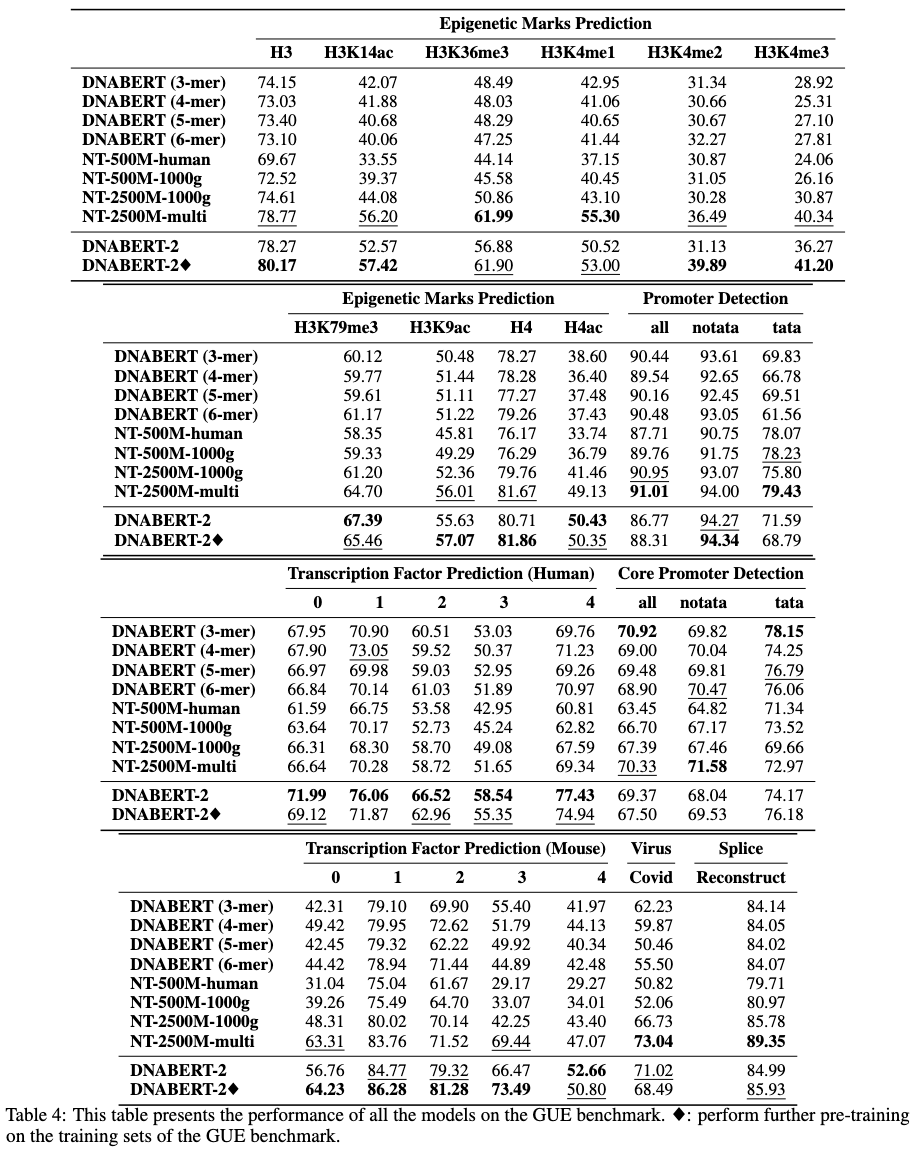
Gue는 게놈 이해를위한 포괄적 인 벤치 마크입니다.
# create and activate virtual python environment
conda create -n dna python=3.8
conda activate dna
# (optional if you would like to use flash attention)
# install triton from source
git clone https://github.com/openai/triton.git;
cd triton/python;
pip install cmake; # build-time dependency
pip install -e .
# install required packages
python3 -m pip install -r requirements.txt
우리의 모델은 Transformers 패키지와 함께 사용하기 쉽습니다.
Huggingface (버전 4.28)에서 모델을로드하려면 :
import torch
from transformers import AutoTokenizer , AutoModel
tokenizer = AutoTokenizer . from_pretrained ( "zhihan1996/DNABERT-2-117M" , trust_remote_code = True )
model = AutoModel . from_pretrained ( "zhihan1996/DNABERT-2-117M" , trust_remote_code = True )Huggingface (버전> 4.28)에서 모델을로드하려면 :
from transformers . models . bert . configuration_bert import BertConfig
config = BertConfig . from_pretrained ( "zhihan1996/DNABERT-2-117M" )
model = AutoModel . from_pretrained ( "zhihan1996/DNABERT-2-117M" , trust_remote_code = True , config = config )DNA 서열의 임베딩을 계산합니다
dna = "ACGTAGCATCGGATCTATCTATCGACACTTGGTTATCGATCTACGAGCATCTCGTTAGC"
inputs = tokenizer(dna, return_tensors = 'pt')["input_ids"]
hidden_states = model(inputs)[0] # [1, sequence_length, 768]
# embedding with mean pooling
embedding_mean = torch.mean(hidden_states[0], dim=0)
print(embedding_mean.shape) # expect to be 768
# embedding with max pooling
embedding_max = torch.max(hidden_states[0], dim=0)[0]
print(embedding_max.shape) # expect to be 768
우리는 dnabert-2 https://github.com/mosaicml/examples/tree/main/examples/benchmarks/bert에 대한 Mosaicbert 구현을 사용하고 약간 수정했습니다. 지침에 따라 모델 교육을 복제 할 수 있어야합니다.
또는 https://github.com/huggingface/transformers/tree/main/examples/pytorch/language-modeling에서 run_mlm.py를 사용할 수 있습니다. 매우 유사한 모델을 생성해야합니다.
교육 데이터는 여기에서 사용할 수 있습니다.
먼저 여기에서 gue 데이터 세트를 다운로드하십시오. 그런 다음 스크립트를 실행하여 모든 작업을 평가하십시오.
현재 스크립트는 4 GPU에 대한 교육을 위해 DataParallel 사용하도록 설정되었습니다. gpus 수가 다른 경우 per_device_train_batch_size 및 gradient_accumulation_steps 변경하여 글로벌 배치 크기를 32로 조정하여 종이의 결과를 복제하십시오. 분산 멀티 GPU 교육 (예 : DistributedDataParallel )을 수행하려면 python torchrun --nproc_per_node ${n_gpu} 으로 변경합니다.
export DATA_PATH=/path/to/GUE #(e.g., /home/user)
cd finetune
# Evaluate DNABERT-2 on GUE
sh scripts/run_dnabert2.sh DATA_PATH
# Evaluate DNABERT (e.g., DNABERT with 3-mer) on GUE
# 3 for 3-mer, 4 for 4-mer, 5 for 5-mer, 6 for 6-mer
sh scripts/run_dnabert1.sh DATA_PATH 3
# Evaluate Nucleotide Transformers on GUE
# 0 for 500m-1000g, 1 for 500m-human-ref, 2 for 2.5b-1000g, 3 for 2.5b-multi-species
sh scripts/run_nt.sh DATA_PATH 0
여기서 우리는 자신의 데이터 세트에서 미세 조정 dnabert2의 예를 제공합니다.
먼저 데이터 세트에서 3 개의 csv 파일을 생성하십시오 : train.csv , dev.csv 및 test.csv . 교육 과정 에서이 모델은 train.csv 에서 교육을 받고 dev.csv 파일에서 평가됩니다. 완료된 경우 교육 후 dev.csv 파일에서 가장 작은 손실로 인한 체크 포인트가로드되어 test.csv 에서 평가됩니다. 유효성 검사 세트가 없으면 dev.csv 및 test.csv 동일하게 만드십시오.
데이터 형식 샘플은 sample_data 폴더를 참조하십시오. 각 파일은 동일한 형식이어야하며 첫 번째 행은 sequence, label 이름의 문서 헤드입니다. 각각의 각 행에는 DNA 서열과 A, A , (예 : ACGTCAGTCAGCGTACGT, 1 )에 의해 연결된 수치 라벨이 포함되어야한다.
그런 다음 다음 코드를 사용하여 자신의 데이터 세트에서 DNABERT-2를 미세 할 수 있습니다.
cd finetune
export DATA_PATH=$path/to/data/folder # e.g., ./sample_data
export MAX_LENGTH=100 # Please set the number as 0.25 * your sequence length.
# e.g., set it as 250 if your DNA sequences have 1000 nucleotide bases
# This is because the tokenized will reduce the sequence length by about 5 times
export LR=3e-5
# Training use DataParallel
python train.py
--model_name_or_path zhihan1996/DNABERT-2-117M
--data_path ${DATA_PATH}
--kmer -1
--run_name DNABERT2_${DATA_PATH}
--model_max_length ${MAX_LENGTH}
--per_device_train_batch_size 8
--per_device_eval_batch_size 16
--gradient_accumulation_steps 1
--learning_rate ${LR}
--num_train_epochs 5
--fp16
--save_steps 200
--output_dir output/dnabert2
--evaluation_strategy steps
--eval_steps 200
--warmup_steps 50
--logging_steps 100
--overwrite_output_dir True
--log_level info
--find_unused_parameters False
# Training use DistributedDataParallel (more efficient)
export num_gpu=4 # please change the value based on your setup
torchrun --nproc_per_node=${num_gpu} train.py
--model_name_or_path zhihan1996/DNABERT-2-117M
--data_path ${DATA_PATH}
--kmer -1
--run_name DNABERT2_${DATA_PATH}
--model_max_length ${MAX_LENGTH}
--per_device_train_batch_size 8
--per_device_eval_batch_size 16
--gradient_accumulation_steps 1
--learning_rate ${LR}
--num_train_epochs 5
--fp16
--save_steps 200
--output_dir output/dnabert2
--evaluation_strategy steps
--eval_steps 200
--warmup_steps 50
--logging_steps 100
--overwrite_output_dir True
--log_level info
--find_unused_parameters False
당사 논문이나 코드에 관한 질문이 있으시면 문제를 시작하거나 Zhihan Zhou ([email protected])를 이메일로 보내주십시오.
작업에서 DNABERT-2를 사용하는 경우 신문을 친절하게 인용하십시오.
dnabert-2
@misc{zhou2023dnabert2,
title={DNABERT-2: Efficient Foundation Model and Benchmark For Multi-Species Genome},
author={Zhihan Zhou and Yanrong Ji and Weijian Li and Pratik Dutta and Ramana Davuluri and Han Liu},
year={2023},
eprint={2306.15006},
archivePrefix={arXiv},
primaryClass={q-bio.GN}
}
dnabert
@article{ji2021dnabert,
author = {Ji, Yanrong and Zhou, Zhihan and Liu, Han and Davuluri, Ramana V},
title = "{DNABERT: pre-trained Bidirectional Encoder Representations from Transformers model for DNA-language in genome}",
journal = {Bioinformatics},
volume = {37},
number = {15},
pages = {2112-2120},
year = {2021},
month = {02},
issn = {1367-4803},
doi = {10.1093/bioinformatics/btab083},
url = {https://doi.org/10.1093/bioinformatics/btab083},
eprint = {https://academic.oup.com/bioinformatics/article-pdf/37/15/2112/50578892/btab083.pdf},
}


