virHEAT
v.0.7.3

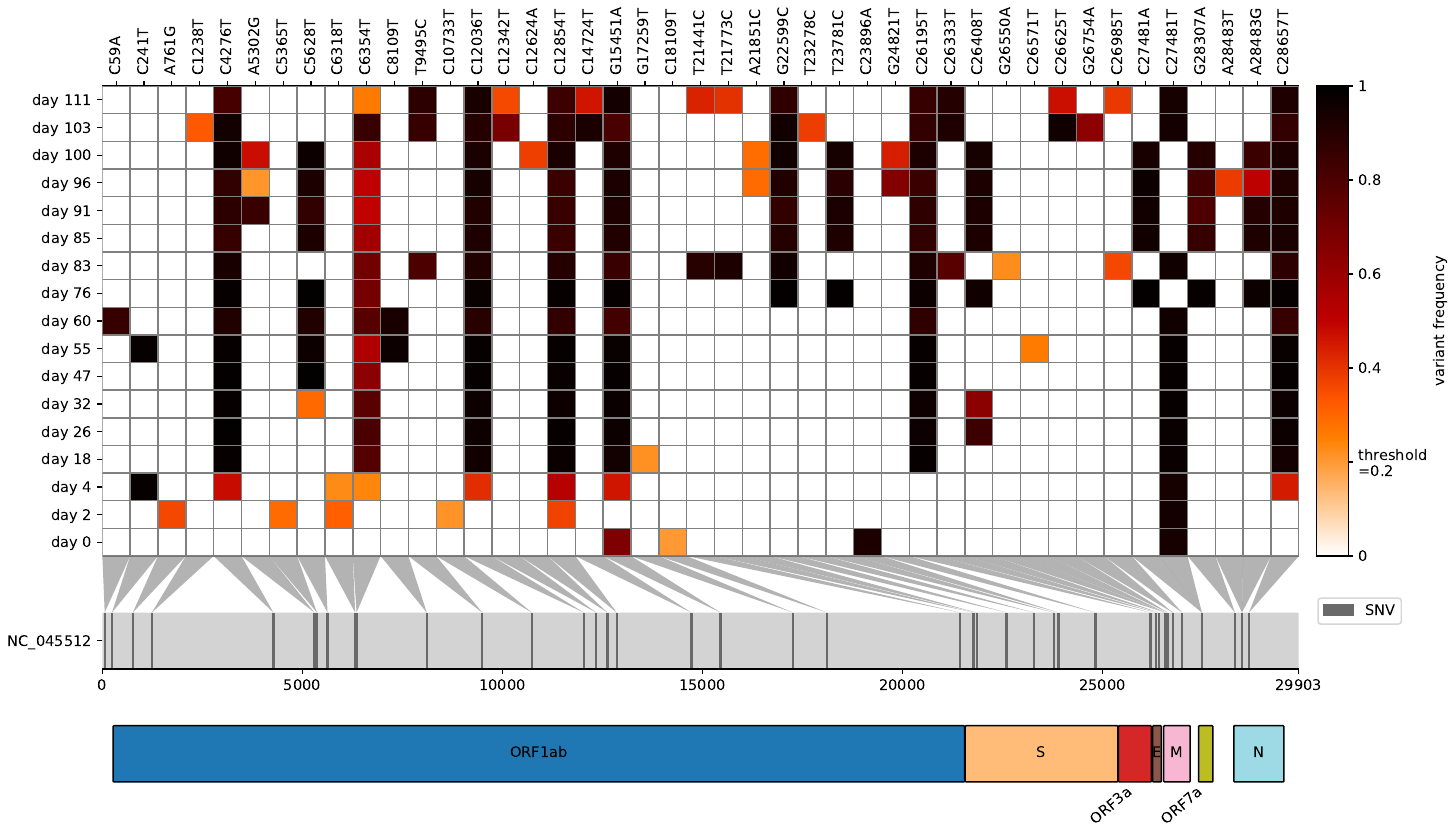
VirheAt는 VCFS를 열 맵으로 시각화하고 각각의 유전자에 대한 MAP 돌연변이를 시각화하는 도구입니다.
원시 판독 값을 바이러스/박테리아 기준 게놈에 매핑 한 후 변형 주파수를 응축하고 동시에 여러 VCF 파일을 비교하고 싶었던 적이 있습니까? Virheat보다 당신을위한 것보다. 당신은 히트 맵을 시각화 할뿐만 아니라 돌연변이를 보유한 유전자를 표시 할 수있는 GFF3 파일로도 읽을 수 있습니다. 이 가벼운 스크립트는 Snipit과 내 변형 주파수 플롯에서 영감을 얻어 두 가지 모두의 최상의 시각화 기능을 얻었습니다.
--scores 
pip install virheatconda install -c bioconda virheatgit clone https://github.com/jonas-fuchs/virHEAT
cd virHEAT
pip install -r requirements.txt
# or
pip install .그것은 이미 그랬습니다. 작동하는지 확인하려면 :
virheat -v현재 Virheat 버전이 표시되어야합니다.
usage: virheat < folder containing vcfs > < output dir > -l or -g [additional arguments]
논쟁 :
positional arguments:
input folder containing input files and output folder
options:
-h, --help show this help message and exit
-r ref_id, --reference ref_id
reference identifier
--name virHEAT_plot.pdf
plot name and file type (pdf, png, svg, jpg). Default: virHEAT_plot.pdf
-l None, --genome-length None
length of the genome (needed if gff3 is not provided)
-g None, --gff3-path None
path to gff3 (needed if length is not provided)
-a [gene ...], --gff3-annotations [gene ...]
annotations to display from gff3 file (standard: gene). Multiple possible.
--gene-arrows, --no-gene-arrows
show genes in arrow format (only if the -g argument is provided) (default: False)
-t 0, --threshold 0 display frequencies above this threshold (0-1)
--delete, --no-delete
delete mutations that are present in all samples and their maximum frequency divergence is smaller than 0.5 (default: True)
-n None, --delete-n None
do not show mutations that occur n times or less (default: Do not delete)
-z start stop, --zoom start stop
restrict the plot to a specific genomic region.
--sort, --no-sort sort sample names alphanumerically (default: False)
--min-cov 20 display mutations covered at least x time (only if per base cov tsv files are provided)
-s scores_file pos_col score_col score_name, --scores scores_file pos_col score_col score_name
specify scores to be added to the plot by providing a CSV file containing scores, along with its column for amino-acid positions, its column for scores, and descriptive score names (e.g., expression, binding, antibody escape, etc.).
This option can be used multiple times to include multiple sets of scores.
-v, --version show program's version number and exit
참조 게놈의 길이를 제공하거나 시퀀스 주석을 얻으려면 GFF3 파일을 제공해야합니다.
또한 돌연변이가 충분히 덮여 있고 회색으로 덮인 세포를 표시하는지 분석 할 수도 있습니다. 이를 위해 먼저 Qualimap을 사용하여 각 BAM 파일에 대한 기본 범위 당 TSV 파일을 생성하고 VCF 파일과 동일한 폴더에 제공합니다. VCF 파일과 같은 이름을 알려주십시오.
또한, 추가 점수의 시각화 (예 : 결합 친화력, 발현 수준, 항체 탈출 등)의 시각화를 포함하는 옵션이있다. 이 기능을 활용하려면 -s 또는 -scores 인수를 사용하고 다음과 같은 인수를 제공하십시오. 1) 점수가 포함 된 CSV 파일의 경로; 2) 고전 표기법 (예 : T430Y)에서 돌연변이 위치를 포함하는이 파일의 열의 이름; 3) 점수 자체를 포함하는이 파일의 열 이름; 4) 플롯에서 레이블로 사용될 설명 점수 이름. 다른 인수로 -s 또는 -스코어 옵션을 반복하여 여러 점수 세트를 동시에 포함시킬 수 있습니다. 예를 들어 입력 및 가능한 출력 데이터는 example_data/example_mave_data 폴더에있는 파일을 참조하십시오.
중요한 면책 조항 : 코드는 GPLV3 라이센스에 따라 있습니다. 코드는 보증이 없습니다. 상업성 또는 특정 목적에 대한 적합성에 대한 묵시적 보증조차 없습니다. 이 프로그램은 무료 소프트웨어입니다. Free Software Foundation, 라이센스의 버전 3 또는 이후 버전에서 게시 한 GNU 일반 공개 라이센스의 조건에 따라 재분배 및/또는 수정할 수 있습니다.


