virHEAT
v.0.7.3

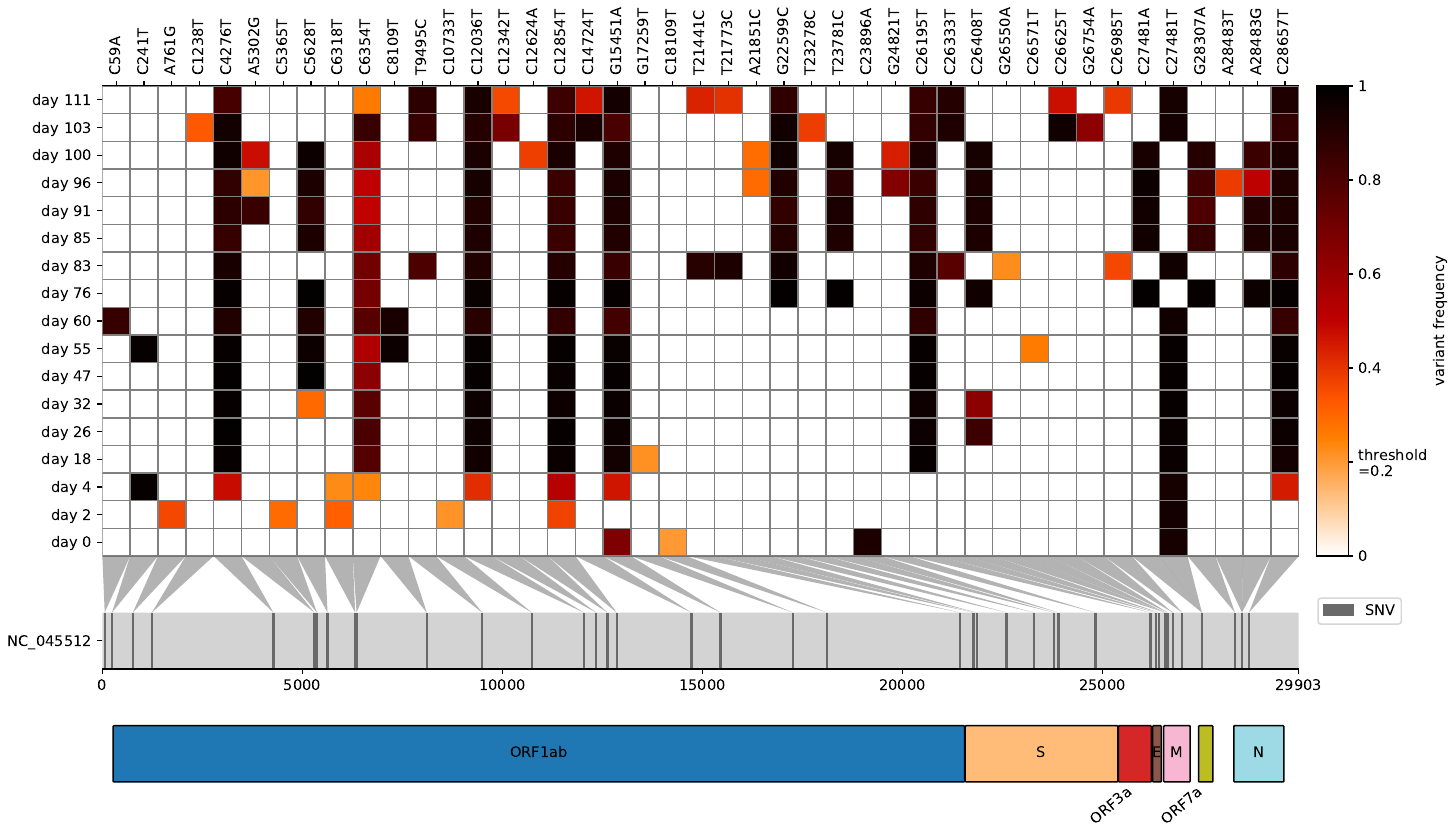
Virheat est un outil pour visualiser les VCF en tant que mutations de carte thermique et cartographier les gènes respectifs.
Vous avez toujours voulu avoir un regard condensé sur les fréquences variantes après la cartographie de vos lectures brutes à un génome de référence virale / bactérienne et comparer plusieurs fichiers VCF en même temps? Que Virheat est pour vous. Vous pouvez non seulement visualiser la carte thermique, mais aussi lire dans un fichier GFF3 qui vous permet d'afficher des gènes hébergeant une mutation. Ce script léger a été inspiré par Snipit et mon tracé de fréquence variant, obtenant les meilleures fonctionnalités de visualisation des deux.
--scores 
pip install virheatconda install -c bioconda virheatgit clone https://github.com/jonas-fuchs/virHEAT
cd virHEAT
pip install -r requirements.txt
# or
pip install .C'était déjà ça. Pour vérifier si cela a fonctionné:
virheat -vVous devriez voir la version Virheat actuelle.
usage: virheat < folder containing vcfs > < output dir > -l or -g [additional arguments]
Arguments:
positional arguments:
input folder containing input files and output folder
options:
-h, --help show this help message and exit
-r ref_id, --reference ref_id
reference identifier
--name virHEAT_plot.pdf
plot name and file type (pdf, png, svg, jpg). Default: virHEAT_plot.pdf
-l None, --genome-length None
length of the genome (needed if gff3 is not provided)
-g None, --gff3-path None
path to gff3 (needed if length is not provided)
-a [gene ...], --gff3-annotations [gene ...]
annotations to display from gff3 file (standard: gene). Multiple possible.
--gene-arrows, --no-gene-arrows
show genes in arrow format (only if the -g argument is provided) (default: False)
-t 0, --threshold 0 display frequencies above this threshold (0-1)
--delete, --no-delete
delete mutations that are present in all samples and their maximum frequency divergence is smaller than 0.5 (default: True)
-n None, --delete-n None
do not show mutations that occur n times or less (default: Do not delete)
-z start stop, --zoom start stop
restrict the plot to a specific genomic region.
--sort, --no-sort sort sample names alphanumerically (default: False)
--min-cov 20 display mutations covered at least x time (only if per base cov tsv files are provided)
-s scores_file pos_col score_col score_name, --scores scores_file pos_col score_col score_name
specify scores to be added to the plot by providing a CSV file containing scores, along with its column for amino-acid positions, its column for scores, and descriptive score names (e.g., expression, binding, antibody escape, etc.).
This option can be used multiple times to include multiple sets of scores.
-v, --version show program's version number and exit
Vous devez soit fournir la longueur de votre génome de référence ou si vous souhaitez obtenir l'annotation de séquence, vous devrez fournir le fichier GFF3.
De plus, vous pouvez également analyser si les mutations sont suffisamment couvertes et afficher des cellules non couvertes en gris. Pour cela, créez d'abord un fichier TSV de couverture par base pour chaque fichier BAM avec Qualimap et fournissez-le dans le même dossier que les fichiers VCF. Donnez-leur le même nom que vos fichiers VCF.
De plus, il existe une option pour inclure des visualisations de scores supplémentaires (par exemple, scores MVE pour l'affinité de liaison, le niveau d'expression, l'évasion des anticorps, etc.) cartographiés aux mutations sur la carte thermique. Pour utiliser cette fonctionnalité, utilisez l'argument -s ou - scores et fournissez les arguments suivants: 1) chemin vers le fichier CSV contenant des scores; 2) le nom de la colonne dans ce fichier contenant des positions de mutation en notation classique (par exemple, T430y); 3) le nom de la colonne de ce fichier contenant les scores eux-mêmes; 4) Un nom de score descriptif qui sera utilisé comme étiquettes dans l'intrigue. Les ensembles de scores multiples peuvent être inclus simultanément en répétant l'option -s ou - scores avec différents arguments. Par exemple, les données de sortie d'entrée et de sortie possibles, veuillez vous référer aux fichiers situés dans le dossier Exemple_Data / Exemple_Mave_Data.
Avertissement important: le code est sous la licence GPLV3. Le code est sans garantie; Sans même la garantie implicite de qualité marchande ou d'adéquation à un usage particulier. Ce programme est un logiciel gratuit: vous pouvez le redistribuer et / ou le modifier en vertu des termes de la licence publique générale GNU publiée par la Free Software Foundation, soit la version 3 de la licence, ou (à votre option) toute version ultérieure.


