virHEAT
v.0.7.3

Virheat ist ein Werkzeug zur Visualisierung von VCFs als Heatmap- und Kartenmutationen in die jeweiligen Gene.
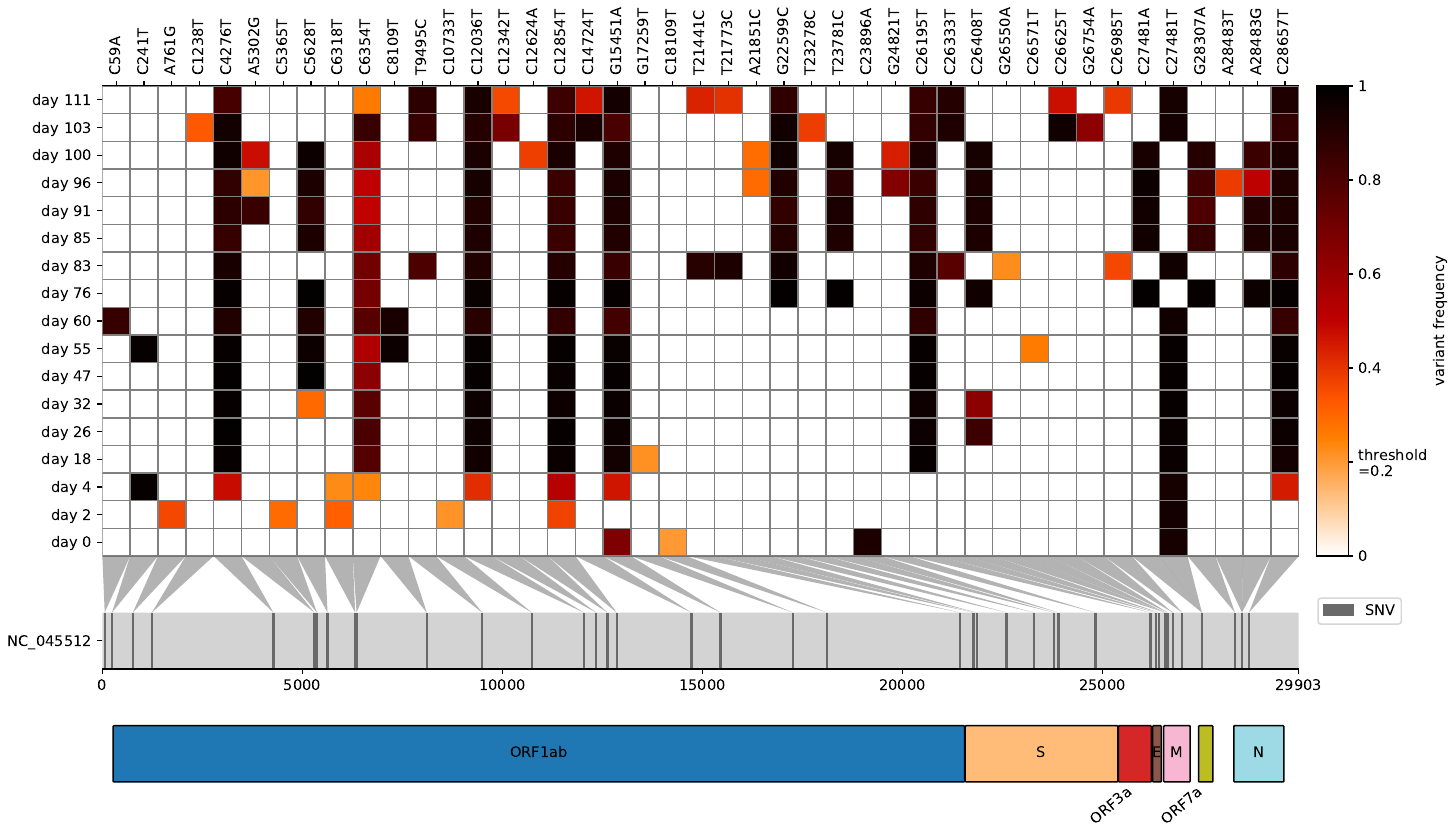
Wollten Sie schon immer einen kondensierten Blick auf die Variantenfrequenzen nach dem Zuordnen Ihrer Rohlesungen auf ein Viral-/Bakterienreferenzgenom und vergleichen gleichzeitig mehrere VCF -Dateien? Als Virheat ist für Sie. Sie können nicht nur die Heatmap visualisieren, sondern auch in einer GFF3 -Datei lesen, in der Sie Gene anzeigen können, die eine Mutation beherbergen. Dieses leichte Drehbuch wurde von Snipit und meinem Variantenfrequenzdiagramm inspiriert, wobei die besten Visualisierungsfunktionen von beidem erhalten wurden.
--scores 
pip install virheatconda install -c bioconda virheatgit clone https://github.com/jonas-fuchs/virHEAT
cd virHEAT
pip install -r requirements.txt
# or
pip install .Das war schon. Um zu überprüfen, ob es funktioniert hat:
virheat -vSie sollten die aktuelle Virheat -Version sehen.
usage: virheat < folder containing vcfs > < output dir > -l or -g [additional arguments]
Argumente:
positional arguments:
input folder containing input files and output folder
options:
-h, --help show this help message and exit
-r ref_id, --reference ref_id
reference identifier
--name virHEAT_plot.pdf
plot name and file type (pdf, png, svg, jpg). Default: virHEAT_plot.pdf
-l None, --genome-length None
length of the genome (needed if gff3 is not provided)
-g None, --gff3-path None
path to gff3 (needed if length is not provided)
-a [gene ...], --gff3-annotations [gene ...]
annotations to display from gff3 file (standard: gene). Multiple possible.
--gene-arrows, --no-gene-arrows
show genes in arrow format (only if the -g argument is provided) (default: False)
-t 0, --threshold 0 display frequencies above this threshold (0-1)
--delete, --no-delete
delete mutations that are present in all samples and their maximum frequency divergence is smaller than 0.5 (default: True)
-n None, --delete-n None
do not show mutations that occur n times or less (default: Do not delete)
-z start stop, --zoom start stop
restrict the plot to a specific genomic region.
--sort, --no-sort sort sample names alphanumerically (default: False)
--min-cov 20 display mutations covered at least x time (only if per base cov tsv files are provided)
-s scores_file pos_col score_col score_name, --scores scores_file pos_col score_col score_name
specify scores to be added to the plot by providing a CSV file containing scores, along with its column for amino-acid positions, its column for scores, and descriptive score names (e.g., expression, binding, antibody escape, etc.).
This option can be used multiple times to include multiple sets of scores.
-v, --version show program's version number and exit
Sie müssen entweder die Länge Ihres Referenzgenoms angeben oder wenn Sie die Sequenzanmerkmale erhalten möchten, müssen Sie die GFF3 -Datei bereitstellen.
Darüber hinaus können Sie auch analysieren, ob Mutationen ausreichend abgedeckt sind und nicht bedeckte Zellen in Grau aufweisen. Erstellen Sie für diese erste TSV -Dateien pro Basisabdeckung für jede BAM -Datei mit Qualimap und geben Sie sie im selben Ordner wie die VCF -Dateien an. Geben Sie ihnen den gleichen Namen wie Ihre VCF -Dateien.
Darüber hinaus besteht die Möglichkeit, Visualisierungen zusätzlicher Bewertungen (z. B. MAVE -Scores für Bindungsaffinität, Expressionsniveau, Antikörper -Flucht usw.) auf Mutationen auf der Heatmap zugeordnet zu haben. Um diese Funktion zu verwenden, verwenden Sie das Argument für -S- oder -Cores und geben Sie die folgenden Argumente an: 1) Pfad zur CSV -Datei, die Bewertungen enthält; 2) der Name der Spalte in dieser Datei, die Mutationspositionen in klassischer Notation enthält (z. B. T430y); 3) der Name der Spalte in dieser Datei, die die Ergebnisse selbst enthält; 4) Ein beschreibender Bewertungsname, der als Etiketten im Diagramm verwendet wird. Mehrere Score -Sets können gleichzeitig aufgenommen werden, indem die Option -s- oder -cores -Option mit unterschiedlichen Argumenten wiederholt wird. Zum Beispiel Eingabe- und mögliche Ausgabedaten finden Sie in den Dateien in Beispiel_Data/Beispiel_Mave_Data.
Wichtiger Haftungsausschluss: Der Code steht unter der GPLV3 -Lizenz. Der Code ist ohne Garantie; Ohne die implizite Garantie für Handelsfähigkeit oder Eignung für einen bestimmten Zweck. Dieses Programm ist kostenlose Software: Sie können es neu verteilt und/oder unter den Bestimmungen der GNU General Public Lizenz wie von der Free Software Foundation, entweder Version 3 der Lizenz veröffentlicht, oder (nach Ihrer Option) jede spätere Version ändern.


