biodiff
biodiff 1.2.1
Alignment Algorithmsを使用してバイナリファイルを比較します。
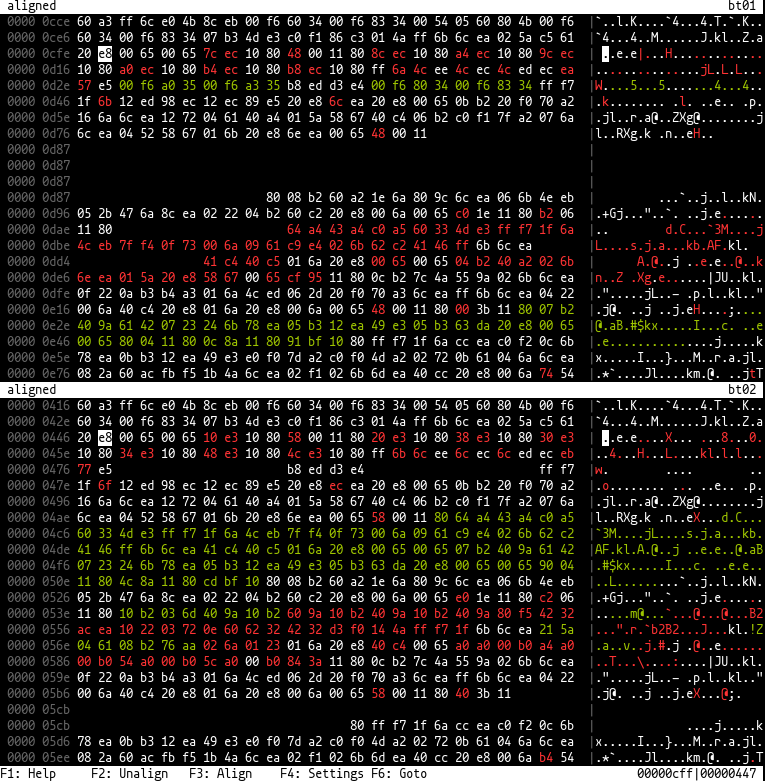
これは、バイナリ拡散のためのツールです。
このツールは、2つのバイナリファイルを並べて表示できるため、同様の場所が両側で同じ位置になり、片側から欠落しているバイトがパッドが付いています。それは、そのために「WFA2」またはrust-bioライブラリ(通常はDNA配列アラインメントに使用される)のバイオインフォマティクスアルゴリズムを使用します。構成用のダイアログボックスは、 cursiveを使用して実行されます。
[ 、 ] 、 0を押すことで調整可能、行ごとの構成可能なバイトbiodiff file_a file_b端末で実行すると、2つのファイルを並べて表示する16進ビューにドロップする必要があります。最初に、ファイルは両側にギャップなしで並べられて表示されません。カーソルとビューを左側と右側が似ている場所に移動し、 F3 (または3 )を押すことで、それらを整列させることができます。これは、標準構成でブロックごとにブロックされます。つまり、カーソルの近くのバイトが最初に並べられ、さらに整列されたブロックが後で両側に表示されます。
また、 F4を使用して設定を変更することにより、グローバルアライメント(ファイル全体の)を実行することも可能です(パラメーターのヘルプを必ず参照してください)。一般的に、2次時間と空間が必要なため、64kbを超えるファイルのグローバルなアライメントはうまく機能しません。また、より高速ですが、精度がわずかに少ない「バンド化された」アルゴリズムもあります。
また、1つのファイルで領域を選択することもできます。F3を押すことにより、Aling Algorithmは、選択したバイトをパターンとして使用して、他のファイルの対応するバイトを見つけるパターンを使用してセミグロールアライメントを行います。
biodiff --print file_a file_bを使用して、DIFFを端末に直接印刷することもできます。その場合(ファイルが時間がかかりすぎないように十分に小さい場合)、 -gglobalフラグを追加してグローバルアライメントを行うことができます(インタラクティブな使用に適したブロックごとのものとは対照的です)。
運が良ければ、プライマリパッケージマネージャーでパッケージを利用できるようになります。Repologyページを参照してください。 [リリース]ページの下にある一部の環境には、ダウンロード可能なバイナリファイルが必要です。または、 cargo install biodiffを使用して、 cargoを使用してこれを設置することもできます。コンパイルするには、 wfa2機能用のCMAKEインストールが必要です。 WINDOWSを使用する場合は、 wfa2サポートが必要な場合はx86_64-unknown-linux-gnuターゲットを使用する必要があることに注意してください。
また、このリポジトリからコードを使用して直接実行することもできます。 cargo run --release -- file_a file_b実行することもできます。構成ファイルは、タグ付きリリース間の互換性があることのみが保証されていることに注意してください。
デフォルトでは、設定はプラットフォーム固有のユーザーディレクトリに保存されます。カスタム設定ディレクトリを使用するには、 BIODIFF_CONFIG_DIR環境変数をbiodiff実行する前に目的のディレクトリパスに設定します。ディレクトリが存在しない場合、自動的に作成されます。
このプロジェクトは、MITライセンスの下でライセンスされています。


