biodiff
biodiff 1.2.1
Comparez les fichiers binaires à l'aide d'algorithmes d'alignement.
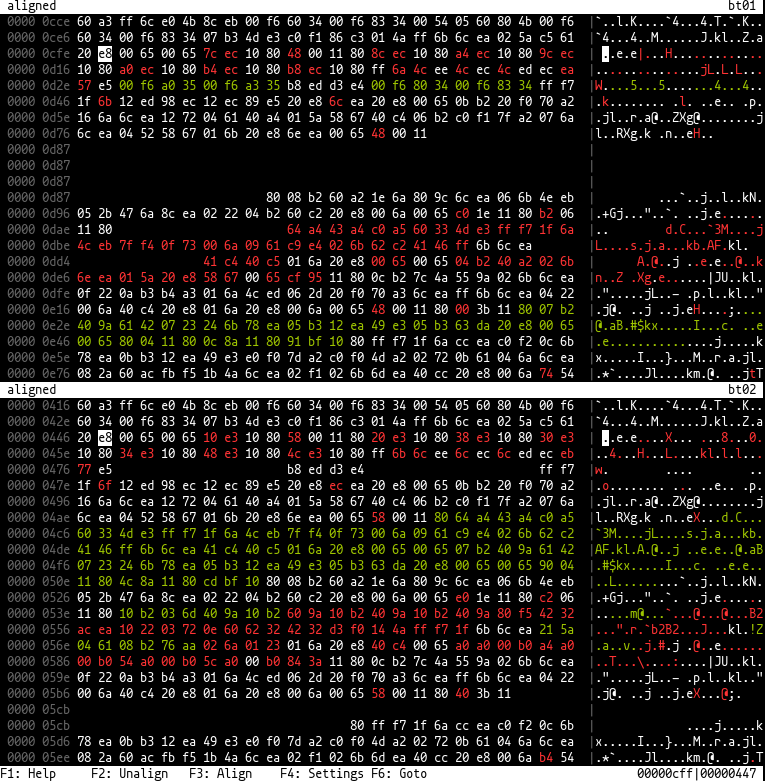
Il s'agit d'un outil de diffusion binaire.
L'outil est capable d'afficher deux fichiers binaires côte à côte afin que les endroits similaires soient à la même position des deux côtés et des octets manquants d'un côté sont rembourrés. Il utilise des algorithmes bio-informatiques de la bibliothèque «WFA2» ou rust-bio (généralement utilisé pour l'alignement de la séquence d'ADN) pour cela. Les boîtes de dialogue pour la configuration sont effectuées en utilisant cursive .
[ , ] , 0 Exécutez biodiff file_a file_b dans un terminal et vous devez être déposé dans une vue hexadécimale affichant deux fichiers côte à côte. Initialement, les fichiers ne seront pas alignés et affichés sans lacunes de chaque côté. En déplaçant le curseur et les vues vers un endroit où le côté gauche et le côté droit sont similaires et appuyant sur F3 (ou 3 ), ils peuvent être alignés. Ceci est fait un bloc par bloc en configuration standard, ce qui signifie que les octets près du curseur sont alignés en premier et d'autres blocs alignés sont affichés plus tard des deux côtés.
Il est également possible de faire l'alignement global (de l'ensemble des fichiers à la fois) en modifiant les paramètres à l'aide de F4 (assurez-vous de consulter l'aide sur les paramètres). Généralement, comme il faut du temps et de l'espace quadratiques, l'alignement global ne fonctionnera pas bien pour les fichiers supérieurs à 64 Ko. Il existe également un algorithme "Banded" qui est plus rapide, mais légèrement moins précis.
Vous pouvez également sélectionner une région sur un fichier et en appuyant sur F3, l'algorithme d'alignement effectuera un alignement semi-bobal à l'aide des octets sélectionnés comme modèle pour trouver les octets correspondants sur l'autre fichier.
Il est également possible d'imprimer le diff directement au terminal en utilisant biodiff --print file_a file_b . Dans ce cas (si les fichiers sont suffisamment petits pour qu'il ne prenne pas trop de temps), vous pouvez ajouter l'indicateur -gglobal pour effectuer un alignement global (par opposition à un blocswise, qui est mieux adapté à une utilisation interactive).
Si vous avez de la chance, un package sera disponible dans votre gestionnaire de packages principal, consultez la page de répologie. Il devrait y avoir des fichiers binaires téléchargeables pour certains environnements sous la page des versions. Alternativement, vous pouvez également l'installer à l'aide cargo en faisant cargo install biodiff . Vous aurez besoin que CMake soit installé pour la fonctionnalité wfa2 à compiler. Notez que dans le cas où vous utilisez Windows, vous devez utiliser la cible x86_64-unknown-linux-gnu si vous souhaitez avoir une prise en charge wfa2 .
Vous pouvez également exécuter directement en utilisant le code à partir de ce référentiel en exécutant cargo run --release -- file_a file_b . Notez que les fichiers de configuration ne sont garantis que pour rester compatibles entre les versions taguées.
Par défaut, les paramètres sont stockés dans un répertoire utilisateur spécifique à la plate-forme. Pour utiliser un répertoire de paramètres personnalisés, définissez la variable BIODIFF_CONFIG_DIR Environment sur le chemin du répertoire souhaité avant d'exécuter biodiff . Si le répertoire n'existe pas, il sera automatiquement créé.
Ce projet est autorisé sous la licence du MIT.


