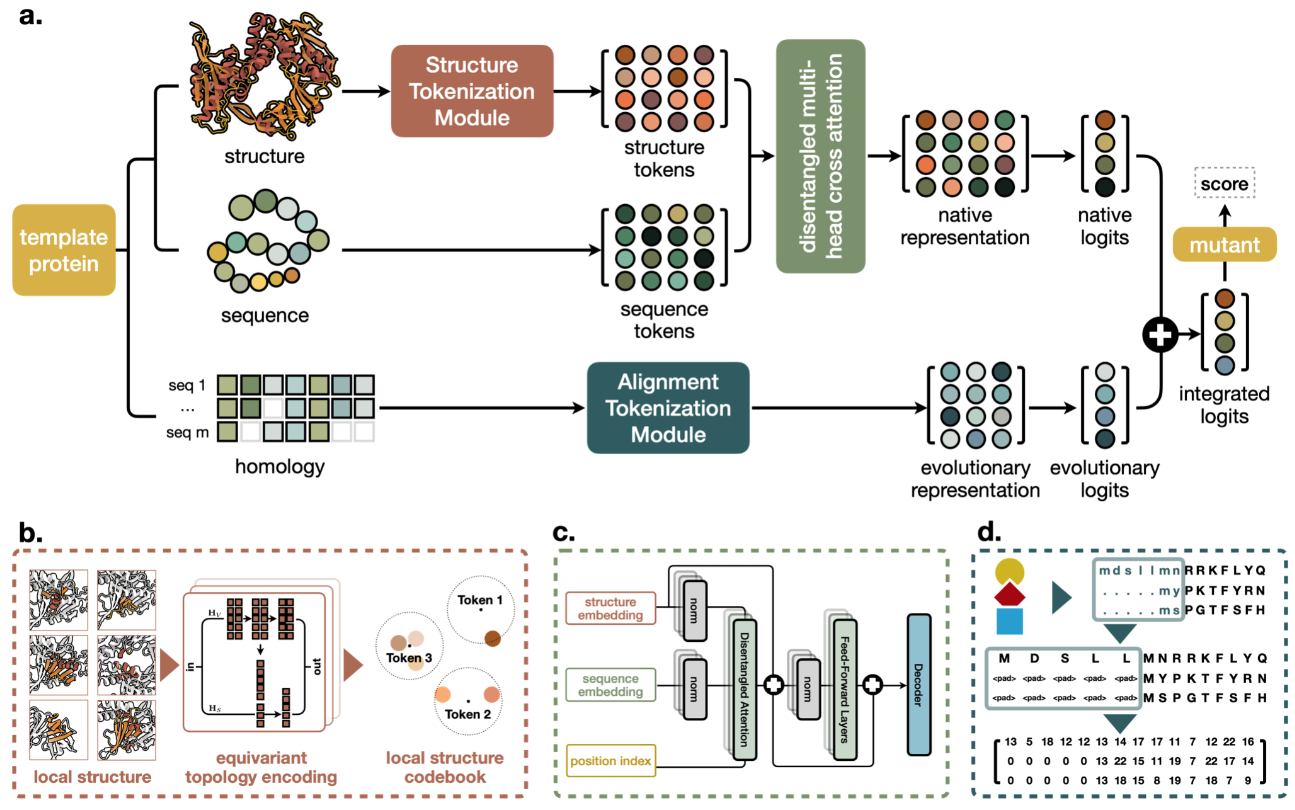
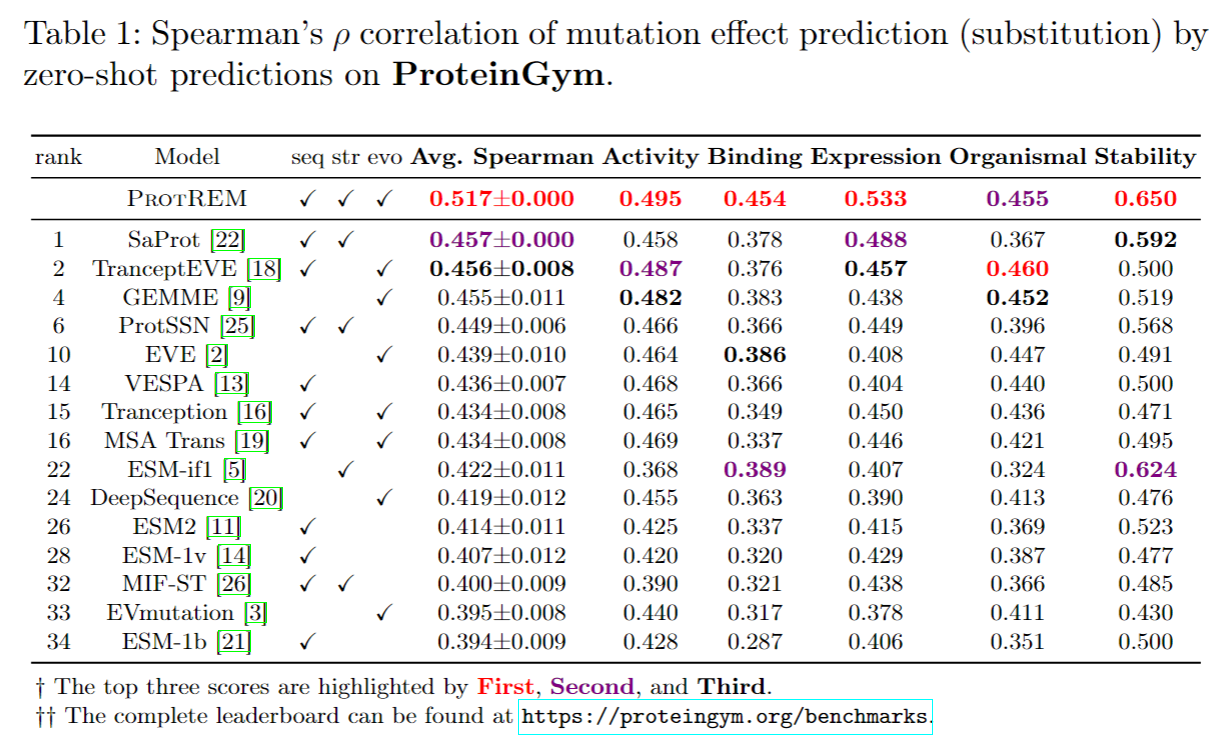
ProREM
1.0.0
請確保您已經安裝了Anaconda3或Miniconda3 。
conda env create -f environment.yml
conda activate prorem
# We need HMMER and EVCouplings for MSA
# pip install hmmer
# pip install https://github.com/debbiemarkslab/EVcouplings/archive/develop.zip
安裝PLMC並更改src/single_config_monomer.txt中的路徑
git clone https://github.com/debbiemarkslab/plmc.git
cd plmc
make all-openmp cd data/proteingym_v1
wget https://huggingface.co/datasets/tyang816/ProREM/blob/main/aa_seq_aln_a2m.tar.gz
# unzip homology files
tar -xzf aa_seq_aln_a2m.tar.gz
# unzip fasta sequence files
tar -xzf aa_seq.tar.gz
# unzip pdb structure files
tar -xzf pdbs.tar.gz
# unzip structure sequence files
tar -xzf struc_seq.tar.gz
# unzip DMS substitution csv files
tar -xzf substitutions.tar.gzprotein_dir=proteingym_v1
python compute_fitness.py
--base_dir data/ $protein_dir
--out_scores_dir result/ $protein_dirdata/ < your_protein_dir_name >
| ——aa_seq # amino acid sequences
| —— | ——protein1.fasta
| —— | ——protein2.fasta
| ——aa_seq_aln_a2m # homology sequences of EVCouplings
| —— | ——protein1.a2m
| —— | ——protein2.a2m
| ——pdbs # structures
| —— | ——protein1.pdb
| —— | ——protein2.pdb
| ——struc_seq # structure sequences
| —— | ——protein1.fasta
| —— | ——protein2.fasta
| ——substitutions # mutant files
| —— | ——protein1.csv
| —— | ——protein2.csv # step 1: search homology sequences
# your protein name, eg. fluorescent_protein
protein_dir= < your_protein_dir_name >
# your protein path, eg. data/fluorescent_protein/aa_seq/GFP.fasta
query_protein_name= < your_protein_name >
protein_path=data/ $protein_dir /aa_seq/ $query_protein_name .fasta
# your uniprot dataset path
database= < your_path > /uniref100.fasta
evcouplings
-P output/ $protein_dir / $query_protein_name
-p $query_protein_name
-s $protein_path
-d $database
-b " 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9 "
-n 5 src/single_config_monomer.txt
# ? Repeat the searching process until all your proteins are done
# step 2: select a2m file
protein_dir= < your_protein_dir_name >
python src/data/select_msa.py
--input_dir output/ $protein_dir
--output_dir data/ $protein_dir 您可以使用Alphafold3服務器,Alphafold數據庫,ESMFOLD和其他工具來獲取結構。
對於濕LAB實驗,請盡可能地獲取高質量的結構。
protein_dir= < your_protein_dir_name >
python src/data/get_struc_seq.py
--pdb_dir data/ $protein_dir /pdbs
--out_dir data/ $protein_dir /struc_seqprotein_dir= < your_protein_dir_name >
python compute_fitness.py
--base_dir data/ $protein_dir
--out_scores_dir result/ $protein_dir您可以使用Protssn(Elife 2024)或Prosst(Neurips 2024)。
答:對於prorem和protssn輸入格式之間的轉換,您可以參考script/data_format_convert.sh 。對於Prosst,JSUT將Alpha更改為0。
protein_dir= < your_protein_dir_name >
python compute_fitness.py
--base_dir data/ $protein_dir
--out_scores_dir result/ $protein_dir
--alpha 0
--model_out_name ProSST-2048答:Protssn使用在氨基酸坐標水平,局部結構上的Prosst模型上使用建模,並明確引入MSA信息。他們每個人在實際的實驗評估中都有自己的優勢和缺點。
如果您使用了我們的代碼或數據,請引用我們的工作。
@article{tan2024prorem,
title={Retrieval-Enhanced Mutation Mastery: Augmenting Zero-Shot Prediction of Protein Language Model},
author={Tan, Yang and Wang, Ruilin and Wu, Banghao and Hong, Liang and Zhou, Bingxin},
journal={arXiv:2410.21127},
year={2024}
}


