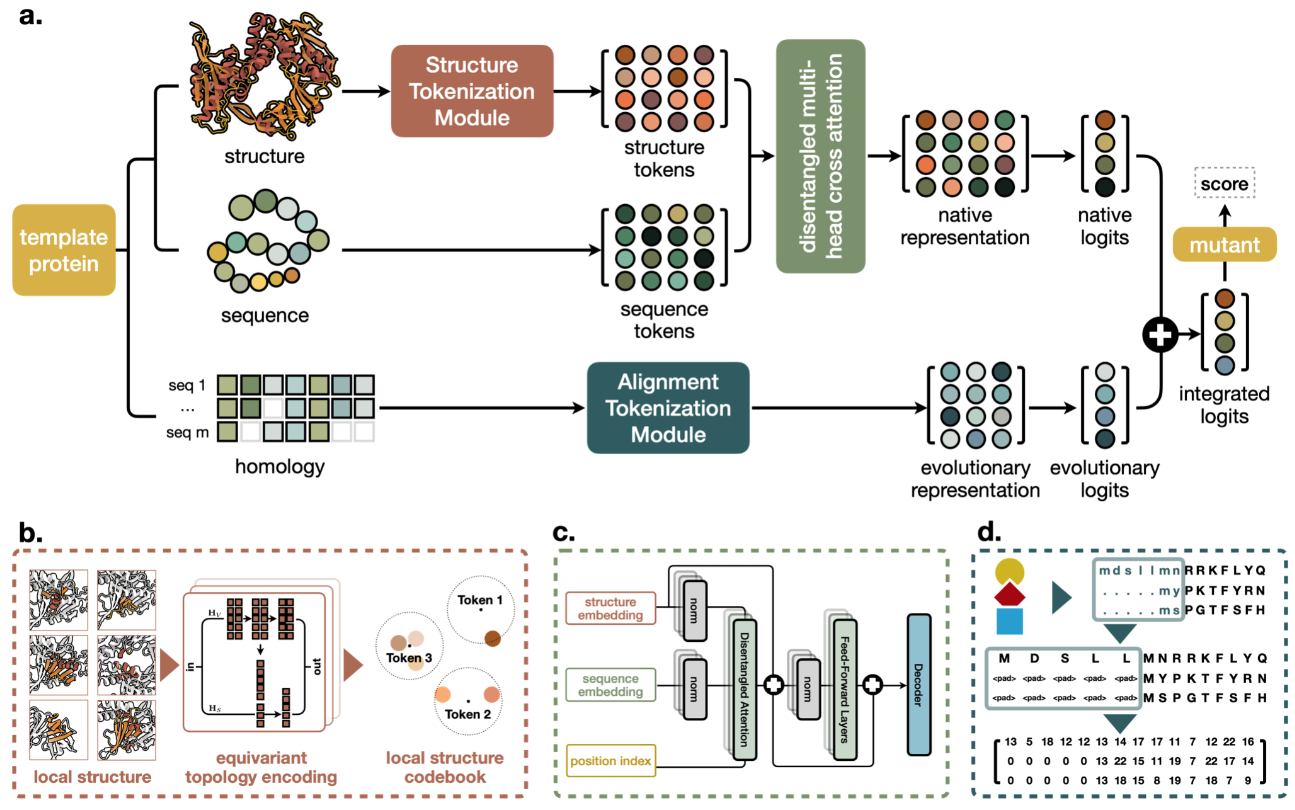
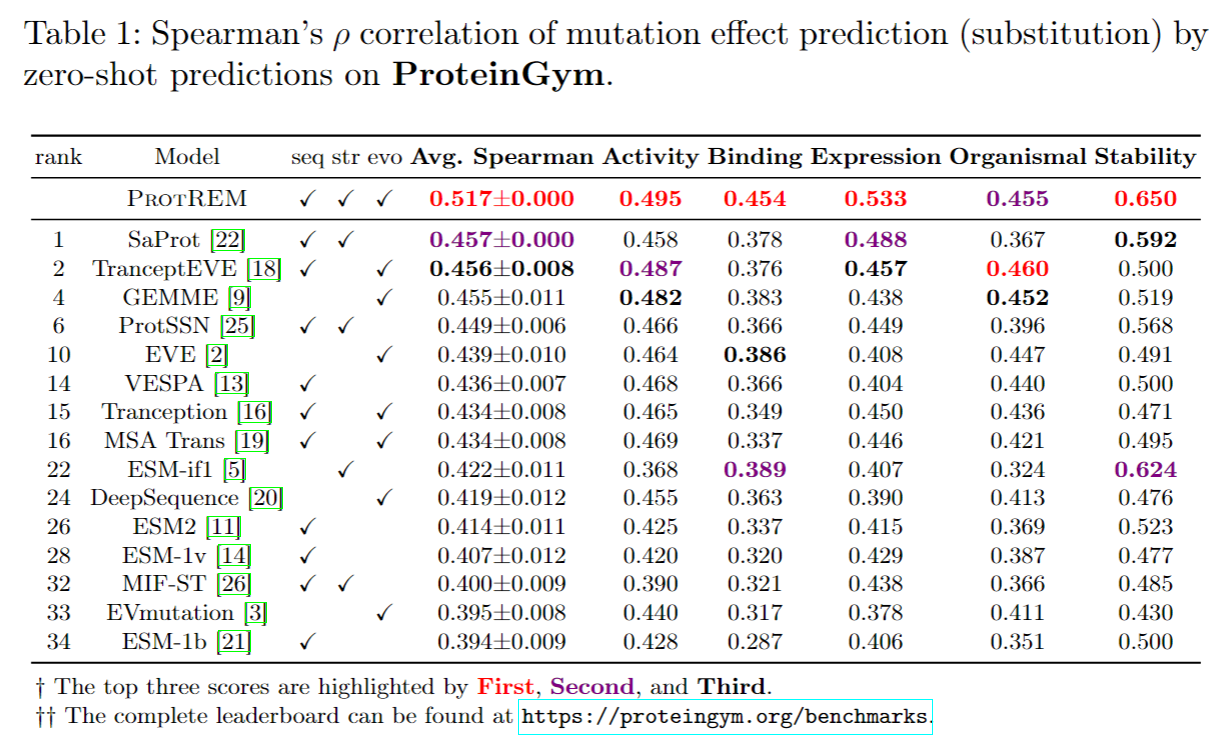
ProREM
1.0.0
Veuillez vous assurer que vous avez installé ANACONDA3 ou MINICONDA3 .
conda env create -f environment.yml
conda activate prorem
# We need HMMER and EVCouplings for MSA
# pip install hmmer
# pip install https://github.com/debbiemarkslab/EVcouplings/archive/develop.zip
Installez PLMC et modifiez le chemin dans src/single_config_monomer.txt
git clone https://github.com/debbiemarkslab/plmc.git
cd plmc
make all-openmp cd data/proteingym_v1
wget https://huggingface.co/datasets/tyang816/ProREM/blob/main/aa_seq_aln_a2m.tar.gz
# unzip homology files
tar -xzf aa_seq_aln_a2m.tar.gz
# unzip fasta sequence files
tar -xzf aa_seq.tar.gz
# unzip pdb structure files
tar -xzf pdbs.tar.gz
# unzip structure sequence files
tar -xzf struc_seq.tar.gz
# unzip DMS substitution csv files
tar -xzf substitutions.tar.gzprotein_dir=proteingym_v1
python compute_fitness.py
--base_dir data/ $protein_dir
--out_scores_dir result/ $protein_dirdata/ < your_protein_dir_name >
| ——aa_seq # amino acid sequences
| —— | ——protein1.fasta
| —— | ——protein2.fasta
| ——aa_seq_aln_a2m # homology sequences of EVCouplings
| —— | ——protein1.a2m
| —— | ——protein2.a2m
| ——pdbs # structures
| —— | ——protein1.pdb
| —— | ——protein2.pdb
| ——struc_seq # structure sequences
| —— | ——protein1.fasta
| —— | ——protein2.fasta
| ——substitutions # mutant files
| —— | ——protein1.csv
| —— | ——protein2.csv # step 1: search homology sequences
# your protein name, eg. fluorescent_protein
protein_dir= < your_protein_dir_name >
# your protein path, eg. data/fluorescent_protein/aa_seq/GFP.fasta
query_protein_name= < your_protein_name >
protein_path=data/ $protein_dir /aa_seq/ $query_protein_name .fasta
# your uniprot dataset path
database= < your_path > /uniref100.fasta
evcouplings
-P output/ $protein_dir / $query_protein_name
-p $query_protein_name
-s $protein_path
-d $database
-b " 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9 "
-n 5 src/single_config_monomer.txt
# ? Repeat the searching process until all your proteins are done
# step 2: select a2m file
protein_dir= < your_protein_dir_name >
python src/data/select_msa.py
--input_dir output/ $protein_dir
--output_dir data/ $protein_dir Vous pouvez utiliser Alphafold3 Server, AlphaFold Database, ESMfold et d'autres outils pour obtenir des structures.
Pour les expériences de laboratoire humide, essayez d'obtenir des structures de haute qualité que possible que possible.
protein_dir= < your_protein_dir_name >
python src/data/get_struc_seq.py
--pdb_dir data/ $protein_dir /pdbs
--out_dir data/ $protein_dir /struc_seqprotein_dir= < your_protein_dir_name >
python compute_fitness.py
--base_dir data/ $protein_dir
--out_scores_dir result/ $protein_dirVous pouvez utiliser ProtSSSN (Elife 2024) ou PROSST (INIPS 2024).
R: Pour la conversion entre le prorém et les formats d'entrée ProtSSSN, vous pouvez vous référer à script/data_format_convert.sh . Pour le prosst, JSUT change l'alpha en 0.
protein_dir= < your_protein_dir_name >
python compute_fitness.py
--base_dir data/ $protein_dir
--out_scores_dir result/ $protein_dir
--alpha 0
--model_out_name ProSST-2048R: ProtSSSN utilise la modélisation au niveau des coordonnées des acides aminés, les modèles PROSST sur la structure locale et le prorème introduit explicitement les informations MSA. Ils ont chacun leurs propres avantages et inconvénients dans une évaluation expérimentale réelle.
Veuillez citer notre travail si vous avez utilisé notre code ou nos données.
@article{tan2024prorem,
title={Retrieval-Enhanced Mutation Mastery: Augmenting Zero-Shot Prediction of Protein Language Model},
author={Tan, Yang and Wang, Ruilin and Wu, Banghao and Hong, Liang and Zhou, Bingxin},
journal={arXiv:2410.21127},
year={2024}
}


