Ligand_Generation
1.0.0
기계 학습 : 과학 기술 (Q1 Journal, 6.8 Impact Factor), 5 권, 2 번 : https://doi.org/10.1088/2632-2153/ad3ee4
사전 프린트 : https://doi.org/10.1101/2023.08.10.552868
기고자 :
@article { Khang Ngo_2024,
doi = { 10.1088/2632-2153/ad3ee4 } ,
url = { https://dx.doi.org/10.1088/2632-2153/ad3ee4 } ,
year = { 2024 } ,
month = { apr } ,
publisher = { IOP Publishing } ,
volume = { 5 } ,
number = { 2 } ,
pages = { 025021 } ,
author = { Nhat Khang Ngo and Truong Son Hy } ,
title = { Multimodal protein representation learning and target-aware variational auto-encoders for protein-binding ligand generation } ,
journal = { Machine Learning: Science and Technology } ,
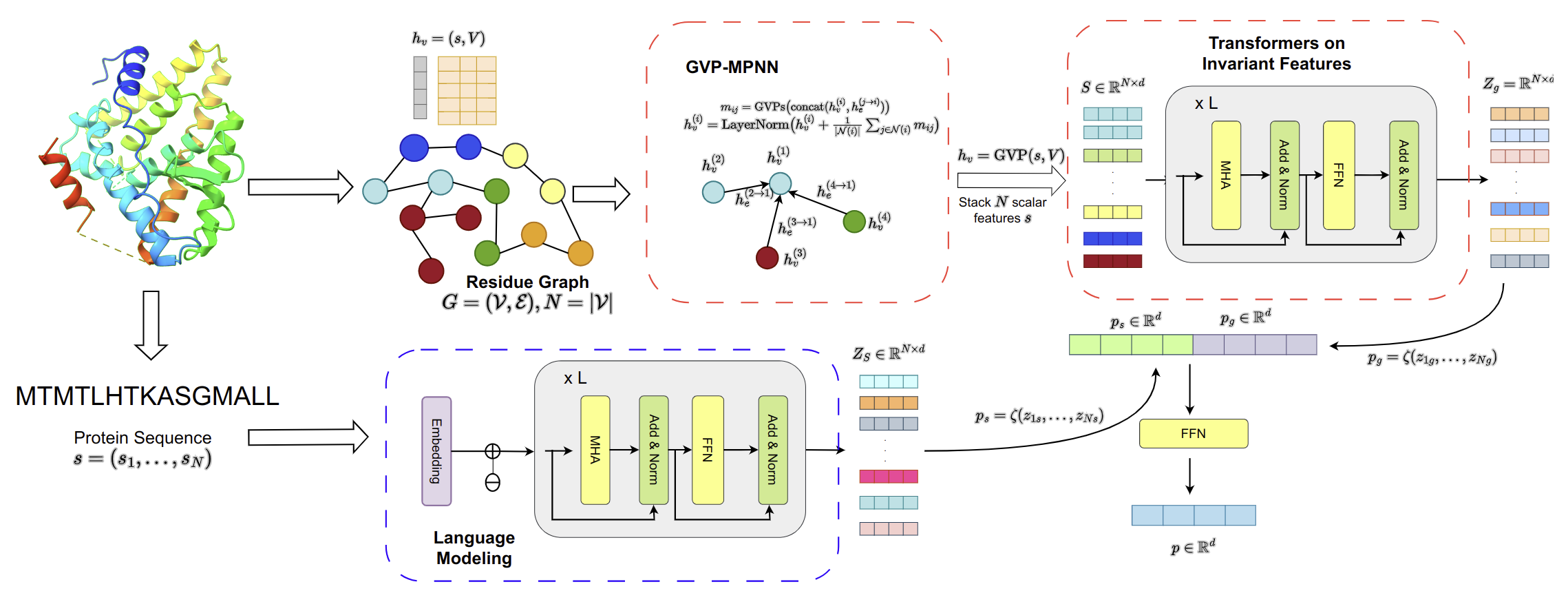
abstract = {Without knowledge of specific pockets, generating ligands based on the global structure of a protein target plays a crucial role in drug discovery as it helps reduce the search space for potential drug-like candidates in the pipeline. However, contemporary methods require optimizing tailored networks for each protein, which is arduous and costly. To address this issue, we introduce TargetVAE, a target-aware variational auto-encoder that generates ligands with desirable properties including high binding affinity and high synthesizability to arbitrary target proteins, guided by a multimodal deep neural network built based on geometric and sequence models, named Protein Multimodal Network (PMN), as the prior for the generative model. PMN unifies different representations of proteins (e.g. primary structure—sequence of amino acids, 3D tertiary structure, and residue-level graph) into a single representation. Our multimodal architecture learns from the entire protein structure and is able to capture their sequential, topological, and geometrical information by utilizing language modeling, graph neural networks, and geometric deep learning. We showcase the superiority of our approach by conducting extensive experiments and evaluations, including predicting protein-ligand binding affinity in the PBDBind v2020 dataset as well as the assessment of generative model quality, ligand generation for unseen targets, and docking score computation. Empirical results demonstrate the promising and competitive performance of our proposed approach. Our software package is publicly available at https://github.com/HySonLab/Ligand_Generation.}
}단백질의 3 차원 구조는 두 개의 압축 파일 인 Prot_3d_for_davis.tar.gz 및 prot_3d_for_kiba.tar.gz에 저장됩니다. 폴더 데이터 로 이동하고 다음 명령을 실행해야합니다.
cd davis
tar –xvzf prot_3d_for_Davis.tar.gz
그리고
cd kiba
tar -xvzf prot_3d_for_kiba.tar.gz
그런 다음 실행할 수 있습니다.
python3 process_protein_3d.py --data {data_name} --device cuda:0
여기에서 data_name은 {kiba, davis}에 있습니다.
당신은 달릴 수 있습니다 :
python3 train_binding_affinity.py --data {data_name} --fold_idx {fold_idx}
data_name은 {kiba, davis}에 있고 fold_idx는 {0,1,2,3,4}에 있어야합니다.
우리는 미리 훈련 된 무조건 VAE에 대한 검문소를 제공합니다. 여기에서 다운로드 할 수 있습니다.
그런 다음 다운로드 된 파일은 Train_vae.py 파일과 동일한 디렉토리에 배치해야합니다.
당신은 달릴 수 있습니다 :
python3 train_vae.py
당신은 달릴 수 있습니다 :
python3 generate_ligand.py --num_mols 100
PDB 파일은 /데이터 디렉토리에 저장해야합니다. 그런 다음 실행할 수 있습니다.
python3 generate_specific_target.py --protein_name [target name]
PDB 파일의 경우 형식은 {target_name} .pdb입니다.
@inproceedings { NEURIPS DATASETS AND BENCHMARKS2021_c45147de,
author = { Townshend, Raphael and V"{o}gele, Martin and Suriana, Patricia and Derry, Alex and Powers, Alexander and Laloudakis, Yianni and Balachandar, Sidhika and Jing, Bowen and Anderson, Brandon and Eismann, Stephan and Kondor, Risi and Altman, Russ and Dror, Ron } ,
booktitle = { Proceedings of the Neural Information Processing Systems Track on Datasets and Benchmarks } ,
editor = { J. Vanschoren and S. Yeung } ,
pages = { } ,
publisher = { Curran } ,
title = { ATOM3D: Tasks on Molecules in Three Dimensions } ,
url = { https://datasets-benchmarks-proceedings.neurips.cc/paper_files/paper/2021/file/c45147dee729311ef5b5c3003946c48f-Paper-round1.pdf } ,
volume = { 1 } ,
year = { 2021 }
} @Article { D3RA00281K ,
author = " Voitsitskyi, Taras and Stratiichuk, Roman and Koleiev, Ihor and Popryho, Leonid and Ostrovsky, Zakhar and Henitsoi, Pavlo and Khropachov, Ivan and Vozniak, Volodymyr and Zhytar, Roman and Nechepurenko, Diana and Yesylevskyy, Semen and Nafiiev, Alan and Starosyla, Serhii " ,
title = " 3DProtDTA: a deep learning model for drug-target affinity prediction based on residue-level protein graphs " ,
journal = " RSC Adv. " ,
year = " 2023 " ,
volume = " 13 " ,
issue = " 15 " ,
pages = " 10261-10272 " ,
publisher = " The Royal Society of Chemistry " ,
doi = " 10.1039/D3RA00281K " ,
url = " http://dx.doi.org/10.1039/D3RA00281K " ,
abstract = " Accurate prediction of the drug-target affinity (DTA) in silico is of critical importance for modern drug discovery. Computational methods of DTA prediction{,} applied in the early stages of drug development{,} are able to speed it up and cut its cost significantly. A wide range of approaches based on machine learning were recently proposed for DTA assessment. The most promising of them are based on deep learning techniques and graph neural networks to encode molecular structures. The recent breakthrough in protein structure prediction made by AlphaFold made an unprecedented amount of proteins without experimentally defined structures accessible for computational DTA prediction. In this work{,} we propose a new deep learning DTA model 3DProtDTA{,} which utilises AlphaFold structure predictions in conjunction with the graph representation of proteins. The model is superior to its rivals on common benchmarking datasets and has potential for further improvement. " }

